Welcome to the March 2015 issue of FREYRFOREWORD!
A monthly round-up of the latest happenings and updates from Freyr.
Happy Reading!
COSMETICS REGIONAL REGULATORY AFFAIRS REGULATION IN THE COSMETIC INDUSTRY
 Cosmetic products have an estimated worth of €67bn in Europe, which is regarded as a massive enterprise. The primary requirement during the development of a cosmetic product is to ensure protection for the user’s health which is also the basis of the cosmetic legislation.
Cosmetic products have an estimated worth of €67bn in Europe, which is regarded as a massive enterprise. The primary requirement during the development of a cosmetic product is to ensure protection for the user’s health which is also the basis of the cosmetic legislation.
PHARMACEUTICAL SERIALIZATION: AN EVOLVING STRATEGY TO ALLEVIATE THE THREAT OF COUNTERFEIT DRUGS
 Counterfeiting, theft, diversion and false returns to manufacturers are few of the problems faced by the pharmaceutical industry across the globe. According to the World Health Organization (WHO), counterfeit drugs make up 1% of the supply in developed countries (including millions of prescriptions in the US alone) and 30-40% in developing countries.
Counterfeiting, theft, diversion and false returns to manufacturers are few of the problems faced by the pharmaceutical industry across the globe. According to the World Health Organization (WHO), counterfeit drugs make up 1% of the supply in developed countries (including millions of prescriptions in the US alone) and 30-40% in developing countries.
IN PERSPECTIVE: AUSTRALIA MOVING TOWARD eCTD SUBMISSIONS
 In 2011-12, the Australian Pharmaceutical industry clocked in exports of $4.06 billion. In the same year, the industry collected around $6.6 billion from Pharmaceutical Benefits Scheme (PBS) sales.
In 2011-12, the Australian Pharmaceutical industry clocked in exports of $4.06 billion. In the same year, the industry collected around $6.6 billion from Pharmaceutical Benefits Scheme (PBS) sales.
CELEBRATING NEW CLIENT WINS
 Our latest new client wins are a fantastic fit for our regulatory expertise and services, we’re absolutely delighted to be working on these projects for one of the Top Global Pharmaceutical Companies.
Our latest new client wins are a fantastic fit for our regulatory expertise and services, we’re absolutely delighted to be working on these projects for one of the Top Global Pharmaceutical Companies.
FREYR SUCCESS STORY – Clinical Cosmetic Safety Assessment
 Learn how Freyr secured significant cost benefits to a Global Top 5, $70+ Bn, Pharma & Consumer Healthcare Company while offering strategic advice on claim support for “to-be” marketed products.
Learn how Freyr secured significant cost benefits to a Global Top 5, $70+ Bn, Pharma & Consumer Healthcare Company while offering strategic advice on claim support for “to-be” marketed products.
FREYR SUCCESS STORY – SSA Quality Variation
 Freyr was engaged by a Global Top 5, $70+ Bn, Pharma & Consumer Healthcare Company for master dossier creation for future markets which incurred 40% savings on cost of compliance.
Freyr was engaged by a Global Top 5, $70+ Bn, Pharma & Consumer Healthcare Company for master dossier creation for future markets which incurred 40% savings on cost of compliance.
 |
| Cosmetic products have an estimated worth of €67bn in Europe, which is regarded as a massive enterprise. The primary requirement during the development of a cosmetic product is to ensure protection for the user’s health which is also the basis of the cosmetic legislation. This protection also enables increased consumer confidence in the brand. |
|
COSMETIC LEGISLATION:
Cosmetics regulation is the main regulatory framework for finished cosmetic products in the EU market. The main aim of the regulation is to ensure protection of consumer’s health and making consumer’s well informed by monitoring the composition and labeling of products and assessment of product safety and mainly focusing on the prohibition of animal testing.The first law governing the manufacture and marketing of safe cosmetic products was introduced in the European Union (EU) in 1976 in the form of a Directive (76/768/EEC). The Cosmetics Regulation, adopted in 2009, replaces Directive 76/768/EC that was adopted in 1976 and had been substantially revised on numerous occasions.Since 11 July 2013, the new EU Regulation 1223/2009 (Cosmetics Regulation) is in force.
WHAT’S NEW IN COSMETIC REGULATION? Manufacturers must follow specific requirements for preparation of a product safety report ahead of placing a product on the market. As per Scientific Committee on Consumer Safety (SCCS) guidelines, a cosmetic product made available on the market shall be safe for human health when used under normal or reasonably foreseeable conditions of use, taking in account like instructions for use and disposal, labeling and any other indication or information provided by the responsible person. COSMETICS PRODUCT SAFETY REPORT: Assessment of safety, which is also deemed as full safety assessment, is to be conducted by a responsible person on the basis of the relevant information. A cosmetic product safety report (CPSR) needs to be generated for every cosmetic product prior to placing the products in the market. In accordance to SCCS guidelines, data on Serious Undesirable Effects (SUE) as well as on any undesirable effect become part of the CPSR. There are two sections in a CPSR, the first section covers “cosmetics safety information” while the second section covers “cosmetics safety assessment”. In addition, No Observed Adverse Effect Level (NOAEL) is the starting point used to calculate the margin of safety (MoS). It is compulsory to state the reasons, if there is no relevant assessment performed. Furthermore, the microbiological quality report and stability test report must also be submitted before safety assessors embark on the final signing of cosmetics safety reports. REPORTING OF SERIOUS UNDESIRABLE EFFECTS: A responsible person will be in charge of notifying Serious Undesirable Effects (SUE) to competent national authorities who will also collect information from users and health professionals.The information will be readily available to share within other EU Member States. NEW RULES FOR THE USE OF NANOMATERIALS IN COSMETIC PRODUCTS: Colorants, preservatives UV-filters and nanomaterials must be explicitly authorized and other nanomaterials present in a product which are not restricted by the Cosmetics Regulation will be the object of a full safety assessment at the EU level. Nanomaterials must be labeled in the ingredients list with the word ‘nano’ in brackets following the name of the substance, e.g. “titanium dioxide (nano)”. INTRODUCTION OF THE NOTION OF ‘RESPONSIBLE PERSON’:
As per SCCS guidelines, prior to placing the cosmetic product on the market the responsible person shall submit, the following information to the Commission by electronic means
COSMETIC PRODUCTS NOTIFICATION PORTAL: It is a centralized notification procedure opted throughout EU for all cosmetic products placed on the EU market. Before placing a product in the market, every manufacturer has to ensure that the product is notified in EU Cosmetic Products Notification Portal (CPNP). It is a free of charge online notification system; CPNP has been created for the implementation of Regulation (EC) No 1223/2009 on cosmetic products. As it is a one-time procedure there is no need for any further notification at the national level in the EU. All information is available electronically to Competent Authorities and Poison Centre’s or similar bodies. PRODUCT INFORMATION FILE: For every cosmetic product (placed on market) there is a Product Information file created and maintained by the responsible person for a period of ten years (following the date on which the last batch of the cosmetic product was placed on the market). ARTWORK AND LABELING: It is mandatory to have listed all ingredients in the cosmetic container labels using identical terms based on the International Nomenclature for Cosmetics Ingredients (INCI) across the whole European Union. Label should include:
FREE MOVEMENT: Member States shall not, refuse, prohibit or restrict cosmetic products availablility on the market which comply with the requirements of regulation. BAN ON ANIMAL TESTING: As per new regulation and guidelines, it is stated as not to have any testing of finished cosmetic products and cosmetic ingredients on animals in the European Union. |
 |
||||
| INTRODUCTION
Counterfeiting, theft, diversion and false returns to manufacturers are few of the problems faced by the pharmaceutical industry across the globe. According to the World Health Organization (WHO), counterfeit drugs make up 1% of the supply in developed countries (including millions of prescriptions in the US alone) and 30-40% in developing countries. By implementing product serialization, counterfeiting by organized crime can be considerably reduced according to pharmaceutical companies and governments of countries across the globe. |
||||
| SERIALIZATION
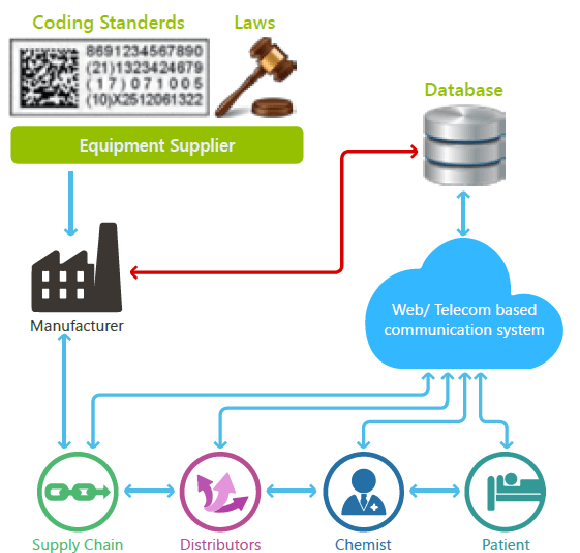
It refers to the allocation and placement of unique markings on a primary package. The markings can be a two-dimensional or RSS bar code, a human readable letter/number code or unique serialized codes that can be written onto a radio-frequency identification (RFID) tag/label. Variable data printers or preprinted labels or cartons are employed to position unique codes on each package which are then read by a vision system. Furthermore, the unique codes are uploaded to an event repository database which can be accessed by various parties, including pharmacists, law enforcement officials and even consumers, once the product is shipped and sold. The individual packages will have unique codes which can be grouped/electronically linked to a shipping case and even to a pallet and other levels of packaging which in turn creates a child/parent/ grandparent relationship. As a result of the grouping, if the bar code on a pallet is scanned at a warehouse, the brand owner or trading partner will have tracking information regarding all shipping containers and primary containers at that warehouse. In addition, once an ePedigree law comes into action, serialization and aggregation will help to track-and-trace products from the point of packaging to the pharmacy or healthcare facility.
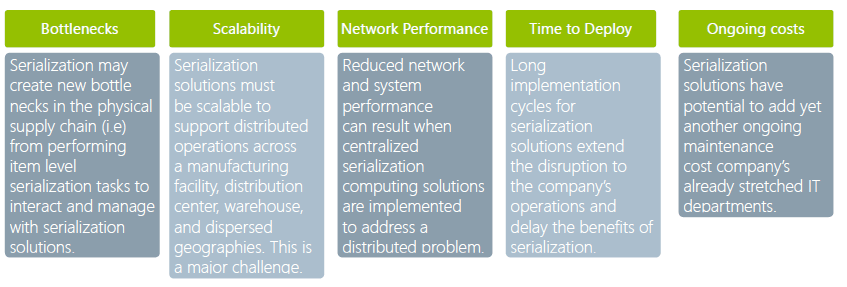
CURRENT SITUATION: Serialization requirements are in heterogeneous stages of development in the US, Canada, EU and in its member nations as well as in Turkey, India, China, Brazil, Argentina and South Korea. Despite the variations found in countries laws, each nation’s regulations tend to be built around GS1 standards and are quite similar. Although the GS1 format is the most desirable standard, International Organization for Standardization (ISO), Internet Engineering Task Force (IETF) and other competing standards also apply to serialization. All activities related to drug serialization that are evolving in different countries are backed up by the overarching global initiative conducted on the World Health Organization (WHO) level. The WHO set up the International Medical Products Anti- Counterfeiting Taskforce (IMPACT). Drug Supply Chain Security Act (DSCSA) Standards for the Interoperable Exchange of Information for Tracing of Certain Human, Finished, Prescription Drugs: How to Exchange Product Tracing Information, released by FDA on 28 November 2014, is meant to address: How information is exchanged between entities within the pharmaceutical supply chain. DSCSA Implementation: Annual Reporting by Prescription Drug Wholesale Distributors and Third-Party Logistics Providers was released on 8 December 2014 and explains how wholesalers and third-party logistics providers (3PLs) should report DSCSA information to FDA on an annual basis. Argentina legislation is effective but limited to certain products; however the number of products falling under this legislation is rapidly growing. In January 2014, ANMAT has published a detailed specification of the central Argentina database (trazabilidat) in Spanish. In December, Brazil published RDC 54/2013, specifying track and trace requirements, accordingly manufacturers must provide serialization and tracking data for three batches of products by 10 December 2015. All pharmaceuticals must be serialized and tracked by 10 December 2016. For a growing market, that’s a critical milestone. On 23 January 2014, the first public hearing discussing implementation was held. Plenty of other countries are preparing for legislation Saudi Arabia by March 2016, Jordan and Ukraine by 2017 including Columbia and Mexico. POSSIBLE STUMBLING BLOCKS IN IMPLEMENTATION
BENEFITS: The leading companies are demanding solutions that will not only please legislative mandates, but also help them in achieving business benefits beyond compliance. These benefits come from the compact product control that serialization provides. They include:
|
||||
| CONCLUSION
In order to meet the legislative mandates for serialization, pharmaceutical companies must start planning now. Most of the governments have been extending compliance deadlines, but leading pharmaceutical companies have already started to pivot on implementing a serialization strategy and defining their requirements on trusted partners with production line serialization solutions. The “wait and see” approach by pharmaceutical companies is not the right way as all pharmaceutical companies will be impacted by serialization regulations.In the future, every pack of drug will be expected to have a unique identifier before going to the market and the industry must make amends to adjust to the changes. |
||||
 |
||
| In 2011-12, the Australian Pharmaceutical industry clocked in exports of $4.06 billion. In the same year, the industry collected around $6.6 billion from Pharmaceutical Benefits Scheme (PBS) sales. A total of 41,000 people had been employed and the industry spent around $1billion on research and development in 2010-11. According to the Complementary Healthcare Council Annual Report (2011-12), sales of complementary medicines stood at $2 billion a year. The Australian Register of Therapeutic Goods (ARTG) shows a total of 48,000 products on its register, of which there are 21,000 devices and 27,000 drugs, of which only 3,500 are registered prescription-only products.
The drugs manufacturing and marketing is regulated by stringent regulations, based on which companies need to submit an application to the regulatory authority for approval.Australia has been accepting paper and non-eCTD Electronic Submissions (NeeS) format of applications. NeeS, is an electronic application however it is way behind the modern type of application, which is implemented in many other parts of the globe, namely eCTD. Australia is now moving forward in an effort to go ahead and implement the eCTD format. In the recent past, Australia has inked an agreement with an eCTD software vendor to receive, review and process electronic (eCTD) applications for the entry of prescription medicines and other therapeutic products on to the ARTG. The nation is working closely with the stake holders and plans to implement this modern format for application by the end of 2014. Regulators at Therapeutic Goods Administration (TGA), will now take a breather owing to this change in the process, however pharmaceutical companies will face a big challenge in moving to this non-familiar format of submissions. Industries will now have to align themselves with various new technical requirements. We might soon witness Australia implementing a more convenient and highly effective format of eCTD. |
 |
||
| As an organization, we at Freyr, have always placed the highest value on our business associations and partnerships. It has been our guiding principle to identify newer opportunities and create exceptional engagement excellence for our clients that transform into long-term relationships. As always, it is a great pleasure to announce the New Wins. | ||
| STRATEGIC REGULATORY SUBMISSIONS SERVICES FOR AN UAE SPECIALTY PHARMA COMPANY:
|
 |
||||||||||||
| HIGHLIGHTS
BUSINESS IMPERATIVES The scope of project is to generate the regulatory and market intelligence data for Pharmaceuticals for seven countries in Sub Saharan Africa region identified by the client – Kenya, Ghana, Tanzania, Zambia, Angola, Nigeria and South Africa. CHALLENGES
CLIENT BENEFITS
FREYR SOLUTION & SERVICES
CLIENT BENEFITS
|
||||||||||||
 |
||||||||||||||
| HIGHLIGHTS
BUSINESS IMPERATIVES
CHALLENGES
CLIENT BENEFITS
FREYR SOLUTION & SERVICES
|
||||||||||||||