Welcome to the August 2015 issue of FREYRFOREWORD!
A monthly round-up of the latest happenings and updates from Freyr.
Happy Reading!
AUSTRALIA REACHES ELECTRONIC ONLY SUBMISSION MILESTONE
 The Australian Therapeutic Goods Administration (TGA) has released the first final regional specification and validation criteria for eCTD format.
The Australian Therapeutic Goods Administration (TGA) has released the first final regional specification and validation criteria for eCTD format.
FIRST ASEAN COUNTRY WITH A NATIONAL eCTD PROGRAM

CELEBRATING NEW CLIENT WINS
 Our latest new client wins are a fantastic fit for our regulatory expertise and services, we’re absolutely delighted to be working on these projects for one of the Top Global Pharmaceutical Companies.
Our latest new client wins are a fantastic fit for our regulatory expertise and services, we’re absolutely delighted to be working on these projects for one of the Top Global Pharmaceutical Companies.
FREYR SUCCESS STORY
 Learn how Freyr helped in securing swift approvals from the Health Authorities for a Canada based Pharmaceutical Company.
Learn how Freyr helped in securing swift approvals from the Health Authorities for a Canada based Pharmaceutical Company.
FREYR SUCCESS STORY
 Learn how Freyr helped a Global Top 5 Pharma and Consumer Health Company in preparing and submitting high quality variation submissions for ‘Type IA’ changes in Finland.
Learn how Freyr helped a Global Top 5 Pharma and Consumer Health Company in preparing and submitting high quality variation submissions for ‘Type IA’ changes in Finland.
 |
||
|
INTRODUCTION
Timelines:
In parallel, the TGA is currently revising the NeeS format to align the corresponding validation criteria with the newly released final eCTD validation criteria. Furthermore additional guidance material for NeeS is being developed and will be released by TGA in the near future. The NeeS format will still be accepted by TGA but a revision of this policy is being considered.A time frame for transitioning from paper to NeeS/eCTD is not determined. TGA has yet to decide the transition period. Once a submission for a product is submitted in the eCTD format, all future submissions for that product must be in the eCTD format in order to take advantage of the lifecycle features of the eCTD standard. When changing from paper or NeeS to eCTD, it is highly recommended but not mandatory to use a baseline as a start of an eCTD. A baseline submission is a compilation of the current status of the dossier, i.e. resubmission of currently valid documents that have already been provided to an agency but in another format. The baseline should normally be submitted as sequence 0000 It should be clearly stated in the cover letter of the 'baseline eCTD sequence' that
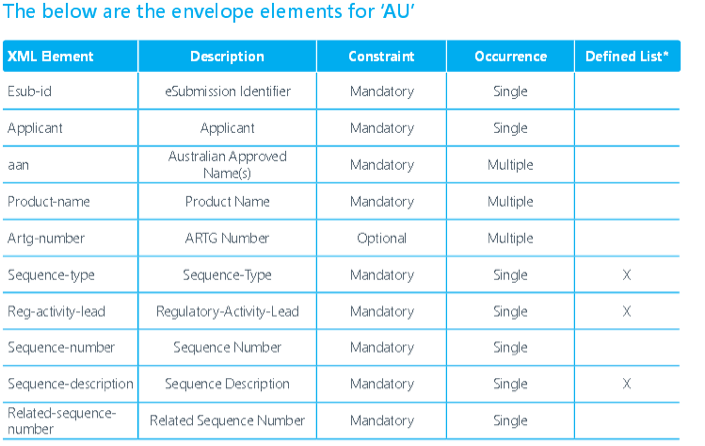
The TGA will not convert new or existing submissions to eCTD format they will only accept validated eCTD submissions published by applicants. TGA is accepting electronic submissions in eCTD and NeeS format. TGA is still accepting regional specification V0.9 but starting from FY 2016 only V3.0 is acceptable. The TGA is planning for RPS (eCTD 4.0).Navigation through an electronic submission is greatly enhanced by the appropriate use of bookmarks and hypertext links. However, the overuse of hyperlinks may confuse rather than help assessors. The maximum size of an individual PDF files used for submission is 100MB. There is no maximum allowable size for the entire submission. The TGA will encourage submission via DVD during the pilot and until they have a fully developed portal facility with capacity for upload. Each eCTD submission should have submission identifier issued by TGA. It is required in the ‘envelope’ information for each submission. Submissions will be referenced by this number and therefore subsequent sequences must include this identifier. To get an eSubmission Identifier, send an email to esubmissions@tga.gov.au (link sends e-mail) with the applicant's name, the Australian Approved Name, and description of the application (trade names, dose form, strength and route of administration). The TGA expects sponsors and applicants to use validation tools to test their own eCTD applications before submitting them to the TGA. A copy of the validation report will need to be attached to the submission. Validation tools are available from software vendors or an example tool can be downloaded from the TGA’s website.TGA will adopt a set of technical validation criteria against which all eCTD and NeeS sequences can be checked. Two categories of validation rules apply: 'Pass/Fail' and 'Best Practice'. The technical validation of an eCTD or NeeS formatted submission is a separate activity to the content validation of a submission and takes place irrespective of the type of the submission. Sequences which fail to meet one or more of the 'Pass/Fail' criteria will be returned to the applicant for correction and resubmission. TGA may accept sequences which fail to meet one or more of the 'Best Practice' criteria; however, the applicant should make every effort to address these areas before the eCTD is submitted to TGA. Submissions which include STFs will be accepted but are not required. However, if STFs are included, they must pass validation. If an FDA submission containing STFs is modified by removing the STFs, the study files must be organized using node extensions.eCTD submissions will be preserved in the TGA's computer network file storage system according to the Australian Government electronic record keeping standards. Applicants should maintain archival copies of submissions according to their business rules. TGA recommends archiving all dossiers in the format the dossier was submitted to the
|
 |
||||
| Thai FDA (Thailand) electronic submission system is to be fully implemented this year. Thai FDA intends to accept dossier in eCTD format: The Drug Regulatory Authority of Thailand (Thai FDA) has initiated the acceptance of Pilot eCTD from October 2014. Formally, eCTD submissions should be accepted from May 2015. | ||||
| Thai eCTD Module
Thai eCTD Module 1 has been developed based on the EU eCTD Module 1 and ICH eCTD specifications v3.2.2 for Module 2 to Module 5. The EU structure is being used as a proven structure and to increase reusability from applications already submitted in the EU region. Thai FDA has released the TH eCTD (Thai eCTD) Module 1 Specifications draft version 0.92 and validation criteria, draft version 0.92. eSubmission and Thailand
Thailand’s Reason’s for Considering eCTD as Global Submission Strategy
Thai eCTD Module:To Improve Reviewing Process
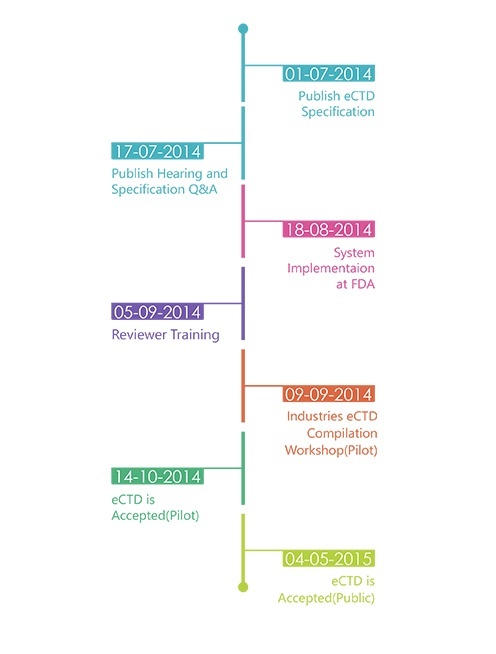
Pilot eCTD
Pilot eCTD: Implementation Timeline
Thai eCTD implementation will have no impact to other or existing applications and variations Filing Dossier in eCTD Format
Validation Report
TH Regional Information
- A cover letter
|
 |
||
| As an organization, we at Freyr, have always placed the highest value on our business associations and partnerships. It has been our guiding principle to identify newer opportunities and create exceptional engagement excellence for our clients that transform into long-term relationships. As always, it is a great pleasure to announce the New Wins of this quarter. | ||
| GLOBAL REGULATORY INTELLIGENCE SERVICES FOR $40+BN, PHARMACEUTICAL COMPANY
STRATEGIC LABELING SERVICES FOR $20+ BN, BIO-PHARMA COMPANY
|
 |
||||||||||||||
| HIGHLIGHTS
BUSINESS HIGHLIGHTS
BUSINESS IMPERATIVES
CHALLENGES The client wanted to urgently meet two-prong objectives:
FREYR SOLUTIONS & SERVICES
CLIENT BENEFITS
|
||||||||||||||
 |
||||||||||
| HIGHLIGHTS
BUSINESS HIGHLIGHTS
BUSINESS IMPERATIVES
CHALLENGES
FREYR SOLUTION & SERVICES
CLIENT BENEFITS
|
||||||||||