Compliance, Audit, and Validation Services - Overview
Are you compliant? This is a question that any pharmaceutical, biotechnology, genomics, chemical, medical device, and supply chain management company finds challenging to answer. These organizations must comply with stringent quality, safety, and compliance requirements in each geography where their products are distributed. The current Regulatory compliance management trend is moving towards global harmonization of quality and safety, emphasizing robust practices. Whether you are a manufacturer or supplier, your market expansion, continual improvement, and customer satisfaction rests principally on the quality standards of your business. Freyr’s Regulatory Compliance, Audit and Validation Center of Excellence (CoE) can help in this effort by assisting you with professionally-aided Compliance, Audit and Validation services for Pharmaceuticals, Medical Devices, Consumer, and healthcare industries such as cGMP (Current Good Manufacturing Practices), GCP (Good Clinical Practice), GDP (Good Distribution Practice), Good Laboratory Practice (GLP), GPVP (Good Pharmacovigilance Practice), and GxP Computer System Validation (CSV). Our pharmaceutical compliance consulting team provides tailored solutions to address your unique compliance challenges.
Freyr provides Regulatory Compliance, Audit, and Validation services that involve evaluating your requirements and suggesting inputs to business processes designed to assure customer satisfaction. With our compliance audit report preparation and readiness strategies, we provide compliance and audit (gap analysis) reports, including suggested Corrective and Preventive Actions (CAPA) to avoid potential 483s and compliance audit findings from regulators. Freyr’s pharmaceutical compliance consulting also extends to pharma automation compliance consulting, ensuring your automated systems meet evolving Regulatory expectations. Our comprehensive approach to compliance management ensures that every stage of your process lifecycle is monitored and optimized for Regulatory alignment.
We provide Regulatory compliance and validation services to ensure that all your processes and products are aligned with the applicable quality, information security, and Regulatory compliance audit requirements such as (but need not be limited to) the following:
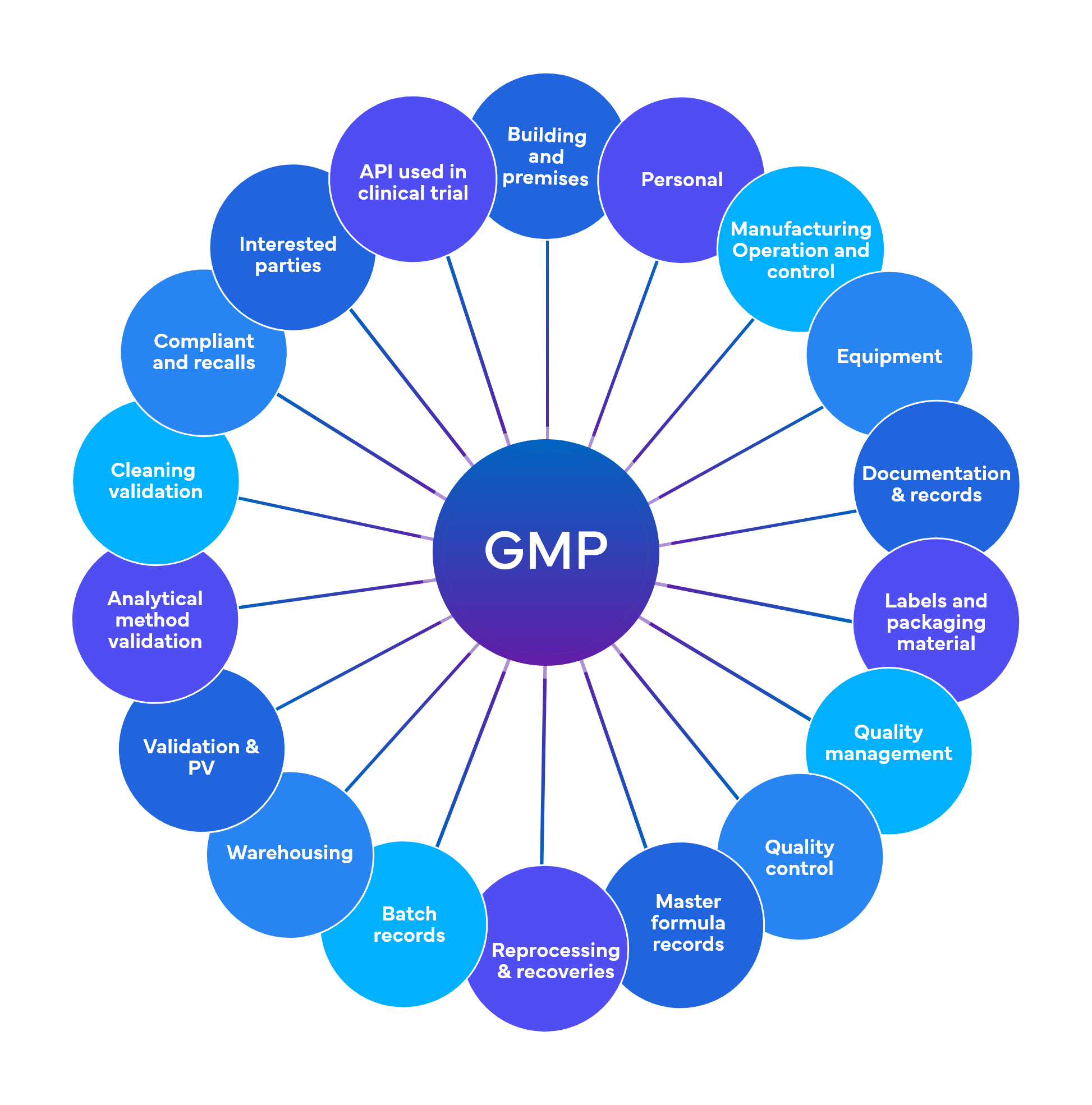
Freyr performs around 700 checks at a preliminary level spread across 18-19 GMP functional areas
Freyr’s Compliance, Audit and Validation
Compliance, Audit, and Validation Services
Quality and Regulatory Compliance – Current Good Manufacturing Practices (cGMP)
- ISO / IEC 17025: 2017 - General Requirements for the Competence of Testing and Calibration Laboratories
- OECD (Organization for Economic Co-operation and Development) - Guidelines of Good Laboratory Practice
- EURACHEM / CITAC Guide: Guide to Quality in Analytical Chemistry - An Aid to Accreditation, 3rd Edition (2016), should be followed in analytical procedures
- 21 CFR 58: Good Laboratory Practice for Non-Clinical Laboratory Studies
- Guide to UK GLP Regulations, Feb 2000 (by GLPMA)
- WHO Handbook on Good Laboratory Practice (GLP): Quality Practices for Regulated Non-Clinical Research and Development
- Health Canada: Finalized Guidance Document - Non-Clinical Laboratory Study Data Supporting Drug Product Applications and Submissions: Adherence to Good Laboratory Practice
- OSHA (Occupational Safety and Health Administration) Standards
- Applicable ICH Guidelines
Good Laboratory Practice
- ISO / IEC 17025: 2017 - General Requirements for the Competence of Testing and Calibration Laboratories
- OECD (Organization for Economic Co-operation and Development) - Guidelines of Good Laboratory Practice
- EURACHEM / CITAC Guide: Guide to Quality in Analytical Chemistry - An Aid to Accreditation, 3rd Edition (2016), should be followed in analytical procedures
- 21 CFR 58: Good Laboratory Practice for Non-Clinical Laboratory Studies
- Guide to UK GLP Regulations, Feb 2000 (by GLPMA)
- WHO Handbook on Good Laboratory Practice (GLP): Quality Practices for Regulated Non-Clinical Research and Development
- Health Canada: Finalized Guidance Document - Non-Clinical Laboratory Study Data Supporting Drug Product Applications and Submissions: Adherence to Good Laboratory Practice
- OSHA (Occupational Safety and Health Administration) standards
- Applicable ICH Guidelines
Good Clinical Practice (GCP) and Good Pharmacovigilance Practice (GVP)
- ICH Harmonized Tripartite Guideline: Guideline for Good Clinical Practice - E6
- E2B(R3) Individual Case Safety Report (ICSR) Specification and Related Files
- E2C(R2) Periodic Benefit-Risk Evaluation Report (PBRER)
- Other Applicable ICH Guidelines
- Guidelines on Good Pharmacovigilance Practices (GVP) Modules
- FDA Safety Reporting Requirements for INDs and BA/BE Studies
- Post Marketing Safety Reporting for Human Drug and Biological Products Including Vaccines
- FDA Regional Implementation Specifications for ICH E2B(R3) Reporting to the FDA Adverse Event Reporting System (FAERS)
- Providing Submissions in Electronic Format – Post Market Non-Expedited ICSRs Technical Questions and Answers
- E2E Pharmacovigilance Planning
Information Security, Data Privacy and Computer System Validation (CSV)
- ISO 27001: 2013 Information Technology – Security Techniques – Information Security Management System – Requirements
- ISO 27002: 2013 Information Technology – Security Techniques – Code of Practice for Information Security Management
- USFDA 21 CFR Part 11: Electronic Records; Electronic Signatures – Scope and Application
- EU GMP Guide Annex 11
- Japanese ERES Guideline Notification No 0401022
- GAMP 5: A Risk-based Approach to Compliant GxP Computerized Systems
- USFDA – Software Validation Guidelines
- Applicable ICH QSEM Guidelines
- Plan and create SOPs and control manufacturing facilities
- Integrated process model
- Maintain brand image
- Risk assessment services
- Overall cost savings
- Implement effective and compliant business processes
- Establishing validation and qualification strategies
- Regulatory and audit compliance expertise
- Expert compliance auditors
Our quality and compliance consulting approach is designed to deliver actionable insights and sustainable improvements, supporting your business in achieving operational excellence and Regulatory success.


 Compliance and Audit Services
Compliance and Audit Services










