
Clinical evaluation report (CER) for medical devices Overview
Any device intended to be marketed in the European Union (EU) must bear a CE mark. In accordance with the EU MDR 2017/745, the requirements for a Clinical Evaluation Report (CER), including process and data requirements, vary based on the class of a device and are necessary for obtaining CE certification of medical devices. Low-risk class I devices can carry out CE-Self Certification. In contrast, other classes of devices (IIa, IIb, III) must process the CE mark certification through an accredited Notified Body (NB). The manufacturer must submit CE technical file to NB for evaluation and issuance of CE mark approval and issuance of the CE certificate. The Clinical Evaluation Report (CER) for medical devices is required to be submitted along with the CE Technical File to comply with CE marking requirements. The CER should be updated continuously with new information from safety checks, follow-up studies, and risk management.
The Clinical Evaluation Report (CER) for medical devices is one of the reports that shall be submitted along with the CE Technical File for complying with CER requirements.
What is a Clinical Evaluation Report (CER)?
Clinical evaluation report writing includes the assessment and analysis of clinical data pertaining to a Medical Device to verify its clinical safety and performance. The clinical evaluation of medical devices is based on the comprehensive analysis of pre- and post-market clinical data relevant to the intended use. The Clinical Evaluation Report includes data specific to the device, as well as any data relating to devices claimed as equivalent by the manufacturer.
A clinical evaluation report consists of scientific literature and analyzed clinical data that was collected either from a clinical investigation of your device or the results of other studies on substantially equivalent devices, where manufacturers must have full access to the equivalent device’s technical, biological, and clinical data and clearly demonstrate how their device matches in all critical aspects. The CER of a medical device demonstrates that the device achieves its intended purpose without exposing users and patients to further risk.
The EU MDR CER must be updated every year. The frequency of CER updates is risk-based and device-specific. If the device carries significant risks, update at least annually, in case the device is marketed for a significant period and is well established the CER can be updated every 2-5 years. Any design changes made to the device design and any new information from PMS data could trigger an update of the CER Report.
The clinical evaluation of medical devices, as framed in the Clinical Evaluation Report (CER), is based on the factors listed below.
- Scientific literature currently available; and/or
- Clinical investigations made; or
- Whether demonstration of conformity with essential requirements based on clinical data is not deemed appropriate.
Stages of Clinical Evaluation Report (CER) Writing
Referring to the new EU-Medical Devices Regulation (MDR) – 2017/745, there are four (04) different stages to performing a clinical evaluation of Medical Devices to prepare a comprehensive EU MDR Clinical Evaluation Report (CER).
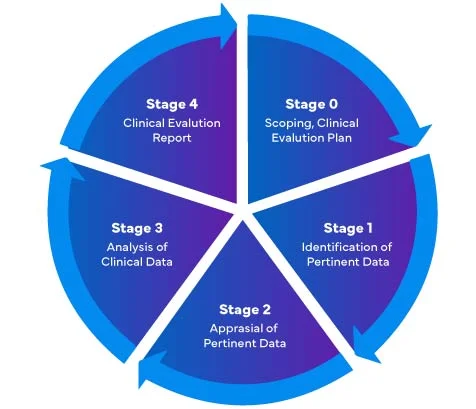
The Medical Device manufacturers entering the EU market for the first time must ensure that their Clinical Evaluation Report is compliant with the EU MDR regulations.
Freyr caters end-to-end CE Certification Services to device manufacturers, including Clinical Evaluation Report writing in line with the newly implemented EU MDR 2017/745 regulations. With strong EU Medical Devices regional expertise, Freyr caters to agency-wise requisites and customizes the Clinical Evaluation Report accordingly.
Clinical evaluation report (CER)
- End-to-end Clinical Evaluation Report writing support, including literature search, as per MEDDEV 2.7/1 revision 4 and the EU Medical Device Regulation (MDR) guidelines.
- Devising a clinical evaluation plan for your organization.
- Identify, search, analyze, and put together the appropriate scientific literature applicable.
- Develop a Clinical Evaluation Report template for your organization.
- Gap Analysis for existing Clinical Evaluation Report.
- DMS tool for your team to collectively contribute to Clinical Evaluation Report writing.
- Integrating PMS Data.
- Develop a standard operating procedure for your team to compile PMS data to update Clinical Evaluation Reports.
- Handling periodic updates of existing Clinical Evaluation Reports, as per the EU MDR guidelines.
- PMS data support for existing devices in the market.
- CE Marking Compliance and CE Marking services.

- Assured compliance with recent applicable regulations.
- Team of qualified clinical expert.
- Cross-functional inputs from Medical Device experts to comply with requirements.
- Full-scope service from compliance, review, and planning.

Frequently Asked Questions (FAQs)
01. What is a Clinical Evaluation Report (CER) and why does it matter?
A Clinical Evaluation Report (CER) is a scientific regulatory document that systematically appraises all available clinical evidence to demonstrate that a medical device is safe, performs as intended, and delivers acceptable clinical benefit-risk outcomes for its intended use under EU MDR. It plays a central role in conformity assessment and ongoing compliance.
02. When should a CER be prepared and updated?
CER preparation begins early in product development as part of a planned clinical evaluation process, and it must be regularly updated throughout the device’s lifecycle whenever new clinical evidence, post-market data, or changes in risk profile emerge, ensuring the benefit-risk assessment remains current.
03. What core elements must a compliant CER include?
A robust CER should reflect a structured appraisal of relevant clinical data, state-of-the-art comparisons, benefit-risk analysis, post-market surveillance inputs, and objective conclusions on conformity with EU MDR safety and performance requirements.
04. How does “state of the art” influence a Clinical Evaluation Report?
“State of the art” is the benchmark of accepted clinical practice and technology against which clinical evidence is compared; it ensures that the device’s benefits and risks are contextualized relative to current standards in medicine, guiding meaningful evidence interpretation.
05. Is a CER required for all medical devices under EU MDR?
Yes, CERs are mandatory for all medical devices marketed in the EU under MDR, regardless of risk class, as they document clinical evidence essential for demonstrating conformity with regulatory safety and performance expectations.
06. What distinguishes a high-quality CER from a basic compliance report?
A high-quality CER integrates comprehensive literature search methodology, clear clinical claims aligned to intended use, rigorous data appraisal, and thoughtful insights on clinical performance and risks, going beyond box-checking to reflect deep understanding of regulatory expectations and clinical context.
07. Why is Freyr considered a leading partner for Clinical Evaluation Report (CER) services?
Freyr is widely recognized for its regulatory-first approach to CER development, combining deep EU MDR expertise, robust clinical evidence appraisal, and lifecycle-driven strategies. Its multidisciplinary teams align clinical, regulatory, and post-market insights to deliver scientifically rigorous CERs that withstand Notified Body scrutiny while supporting long-term compliance.







