
Ensuring Seamless Compliance for UK Medical Device Regulations & Registration
Foreign manufacturers must comply with the UK Medical Devices Regulations 2002 (as amended). All medical devices and IVDs must be registered with the MHRA before sale in the UK. Non-UK manufacturers are required to appoint a UK Responsible Person (UKRP) to represent them. The MHRA lays down strict requirements on documentation, labeling, and vigilance reporting to ensure patient safety and product traceability.
Navigating through the new requirements proposed in the consultation outcome for UK can be complex around UKCA marking, data requirements, and post-market expectations. Many companies face challenges with incomplete technical files, differing device classifications, and limited familiarity with the Device Online Registration System (DORS). Regulatory updates and evolving MHRA guidance further complicate planning and resource allocation.
Freyr simplifies every step of UK market entry. We manage DORS submissions, act as your UKRP, ensure documentation readiness, and bridge compliance gaps during CE-to-UKCA transition. Our regulatory experts guide you through changing MHRA policies, helping you achieve compliant, timely, and cost-efficient market access.
Step-by-Step UK Medical Device Registration Process
Registering a medical device in the UK involves several defined stages. Here’s how Freyr manages the entire process for you:
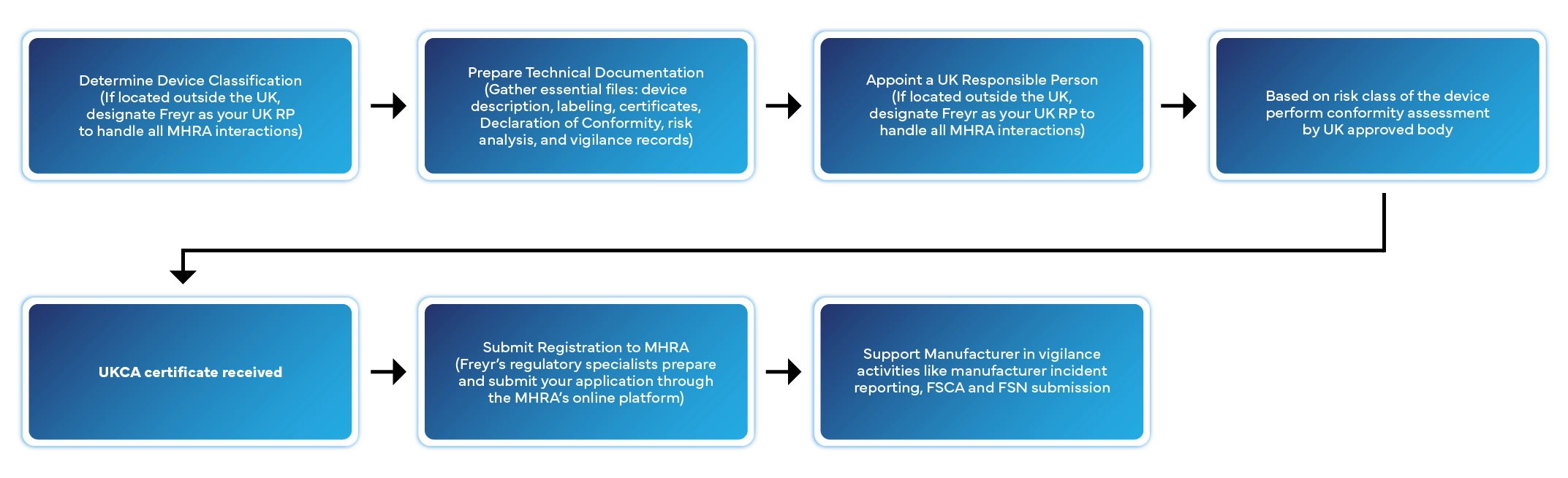
Typical processing time: 2–6 weeks depending on device class and completeness of documentation.
Freyr Medical Device UK Key Offerings

Device Registration Freyr manages complete MHRA registration through the DORS platform, ensuring compliant submissions, accurate data, and timely approvals for all device categories.

UK Responsible Person As your designated UKRP, Freyr represents you to the MHRA, handles all communication, and ensures continuous compliance and vigilance monitoring.

TF / Dossier Compilation Freyr compiles technical files and dossiers that meet UK regulation requirements for UKCA marking, ensuring readiness for audits, inspections, and submissions.

QMS Support We assist in implementing and maintaining ISO 13485-compliant Quality Management Systems tailored to UK MDR and MHRA expectations.

Regulatory Writing Support Freyr provides expert preparation of CERs, PMS plans, PSURs, and risk management documentation, ensuring technical clarity and regulatory accuracy.

Labeling and Compliance Our team ensures your labeling, IFUs, and packaging meet UKCA labeling and language requirements, maintaining consistency and compliance.

Post-Market Surveillance Freyr supports PMS activities including adverse event reporting, vigilance submissions, and MHRA updates to sustain ongoing market access.
Freyr UK Responsible Person (UKRP) Service Offerings
Freyr serves as your designated UKRP, ensuring full compliance with MHRA’s UK Medical Devices Regulations 2002. We represent your company locally in the UK and handle all communication with the MHRA.
- Device Registration with MHRA
Freyr acts as your appointed UK Responsible Person (UKRP) to manage the complete MHRA registration process via the DORS platform ensuring every device is accurately listed, verified, and eligible for sale in the UK. - Documentation & Conformity Assurance
Our regulatory experts ensure that your Declaration of Conformity, technical documentation, and product certifications are available and maintains a copy for making the documentation available to MHRA, upon request. - Responding to MHRA Queries
Freyr directly handles all communications and clarifications with the MHRA on your behalf ensuring timely, accurate responses to regulatory queries or post-market reviews. - Vigilance & Incident Communication
As your UKRP, Freyr serves as the primary liaison for safety-related issues. We coordinate between manufacturers, healthcare professionals, patients, and the MHRA on adverse events, ensuring proper reporting and corrective actions. - Inspection & Audit Readiness
Freyr maintains all required documentation and correspondence for MHRA inspections and audits. Our team ensures that technical files, labeling, and post-market records are readily available.
Book a meeting with our experts today
Why Partner with Freyr?
- End-to-end regulatory expertise that spans from pre-market registration to post-market vigilance, managing every stage of compliance.
- A proven track record with 1500+ device registrations completed successfully across diverse categories.
- Local UK presence with on-ground regulatory specialists in Reading, supported by global delivery teams.
- Tailored transition planning that provides strategic support to move from CE to UKCA smoothly and cost-effectively.
- Transparent communication through direct liaison with MHRA and proactive compliance updates to clients.
- Trusted by global brands, with Freyr serving 470+ manufacturers worldwide as their regulatory partner.
Frequently Asked Questions (FAQs)
01. What is the UK Medical Device Registration process?
The UK Medical Device Registration process involves notifying the MHRA (Medicines and Healthcare products Regulatory Agency) about your device before it is marketed in Great Britain. Manufacturers must provide company details, device classification, and technical documentation. Non-UK manufacturers must also appoint a UK Responsible Person (UK RP) to handle registration and ongoing compliance communications.
02. Who needs to appoint a UK Responsible Person (UK RP)?
Any manufacturer located outside the United Kingdom must appoint a UK Responsible Person before placing devices on the UK market. The UK RP serves as the manufacturer’s regulatory contact, ensuring all technical documentation, declarations, and MHRA communications are properly maintained. UK-based manufacturers can directly interact with the MHRA without a UK RP.
03. What documentation is required for MHRA registration?
The MHRA requires essential documentation, including the Declaration of Conformity, device description and classification, manufacturer details, and labeling information. For higher-risk devices, technical files and clinical evidence may also be reviewed. Having complete, audit-ready documentation ensures faster approvals and smoother post-market inspections by the MHRA or authorized representatives.
04. How long does the MHRA registration process take?
Typically, MHRA registration takes 2 to 6 weeks, depending on the device class, completeness of documentation, and whether a UK Responsible Person is involved. Simple Class I devices can be processed faster, while complex or higher-risk products may take longer due to additional data validation and technical documentation reviews.
05. What is the difference between CE marking and UKCA marking?
CE marking demonstrates compliance with European Union regulations, while UKCA marking (UK Conformity Assessed) applies to devices marketed in Great Britain. Since Brexit, UKCA has replaced CE for the UK market, though CE marking continues to be accepted temporarily. Northern Ireland still recognizes CE and CE UKNI marks under EU alignment rules.
06. What are post-market responsibilities after registration?
After registration, manufacturers and UK Responsible Persons must conduct post-market surveillance, report adverse incidents, update MHRA on product or labeling changes, and renew registrations as needed. They must also maintain technical documentation for at least 10 years. These activities ensure continuous compliance and safeguard patient health throughout the device’s lifecycle.
07. How often should MHRA registration information be updated?
Registration information should be updated promptly whenever there is a change in device classification, labeling, manufacturing site, or responsible entity. The MHRA expects immediate updates from the UK Responsible Person or manufacturer to maintain accurate market records. Regular internal audits help ensure your data remains current and compliant with evolving regulations.
08. What are the penalties for non-compliance with MHRA regulations?
Failure to comply with UK medical device registration or post-market requirements can lead to regulatory warnings, product withdrawal, fines, or prosecution. The MHRA also has authority to suspend or revoke market authorization. Proactive compliance and appointing a qualified UK Responsible Person help manufacturers avoid penalties and maintain market continuity.
09. How does Freyr simplify UK medical device registration?
Freyr provides end-to-end support from acting as your UK Responsible Person (UKRP) to handling MHRA registration, technical documentation, and ongoing compliance. Our experienced consultants streamline the process, reduce approval timelines, and help ensure continuous market access.







