3 min read
BIMO stands for Bioresearch Monitoring, a program of on-site inspection and data audits to monitor all aspects of conduct and reporting of the US Food and Drug Administration (FDA)-regulated research. The program was established in 1977 after a need was identified to audit clinical research sites. The main objective of this program is to ensure the quality and integrity of the submitted data for new product approvals and marketing applications. In addition, this program also protects the rights and welfare of human as well as animal subjects involved in the FDA-regulated research.
The key objectives of the BIMO Program
Annually, over 1000 inspections are conducted. The primary objectives covered under the BIMO program are:
- Audit clinical data
- Inspection of ongoing clinical research
- Inspection of non-clinical laboratories
- Inspection of Institutional Review Boards (IRB)
Which products fall under the purview of BIMO Audit?
The BIMO is applicable for Drugs, Biologics, Medical Devices, Food products, Tobacco products, and Veterinary products. The compliance program is overseen by the FDA’s six (06) product centers - Center for Biologics Evaluation and Research (CBER), Center for Devices and Radiological Health (CDRH), Center for Drug Evaluation and Research (CDER), Center for Food Safety and Applied Nutrition (CFSAN), Center for Tobacco Products (CTP), and Center for Veterinary Medicine (CVM).
Which companies are subjected to BIMO Audit?
Both domestic, as well as international companies carrying out or falling under any of the below activities are subjected to the requirements of Bioresearch Monitoring -
- Non-clinical testing laboratories for Good Laboratory Practice (GLP) compliance
- Clinical investigators for Good Clinical Practice (GCP) compliance
- Sponsors
- Contract Research Organizations (CROs)
- Clinical trial monitors
- In vivo bioequivalence facilities
- Institutional Review Boards (IRBs)
Which compliance programs fall under the BIMO Program?
The US FDA may conduct a BIMO audit at any time through the seven (07) multi-centers compliance programs. These seven multi-centers compliance programs are implemented through –
- Clinical Investigator (CI) and Sponsor Investigator (SI) Inspection
- Institutional Review Board (IRB) Inspection
- Contract Research Organization/Sponsor/Monitor (CRO/S/M) Inspection
- Good Lab Practice (GLP) Inspection
- Bioequivalence-Bioavailability (BEQ) Inspection
- Post-marketing Adverse Drug Experience (PADE) Reporting Inspection
- Risk Evaluation Mitigation and Strategy (REMS) Reporting Inspection
Each of these programs outlines a detailed scope of review or inspection to be conducted to ensure compliance with the FDA.
Which regulations are applicable for BIMO Audit?
The regulations - 21 CFR 50 - Protection of Human Subjects, 21 CFR 54-Financial Disclosure, 21 CFR 56-IRBs, 21 CFR 58-Good Laboratory Practice for non-clinical laboratories, 21 CFR 809-In Vitro Diagnostic Products, and 21 CFR 812-Investigational Device Exemption are applicable for the BIMO Audit.
How many audits are carried out under BIMO Program annually?
The number of BIMO audits conducted by the US FDA varies each year. In recent years, the number of on-site inspections has declined due to the onset of the COVID-19 pandemic, and the FDA had to withhold all the on-site surveillance of the clinical studies. Only specific critical and crucial clinical studies were being monitored.
The “Remote Regulatory Assessments” (RRAs) were introduced during the COVID-19 pandemic to remotely monitor regulated research. RRAs are conducted via video teleconferences and are a voluntary initiative for remotely evaluating the data and processes. Yet the fact to be noted here is that RRAs are not equivalent or an alternative to the on-site inspection but is merely a procedure that evolved due to the COVID-19 pandemic.
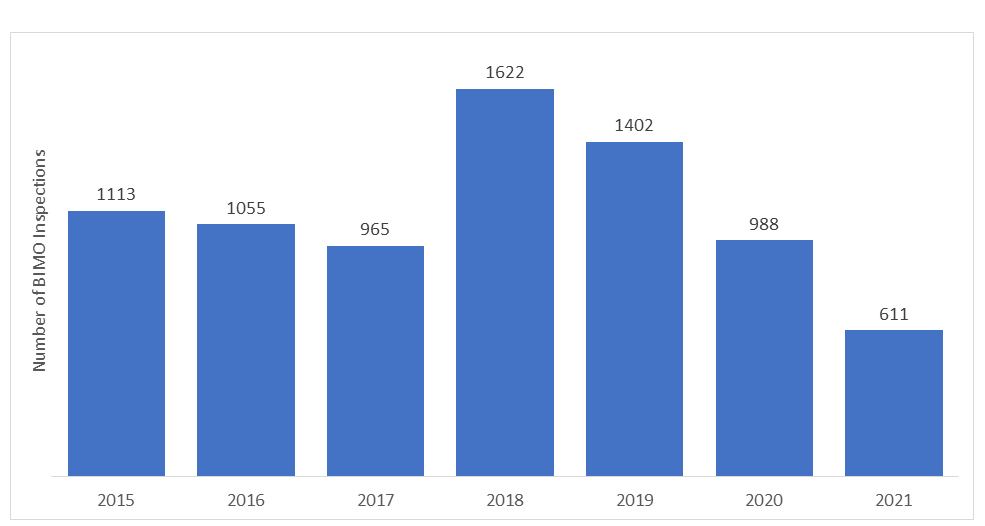
*The data represented for the years 2020 and 2021 do not include RRA inspections
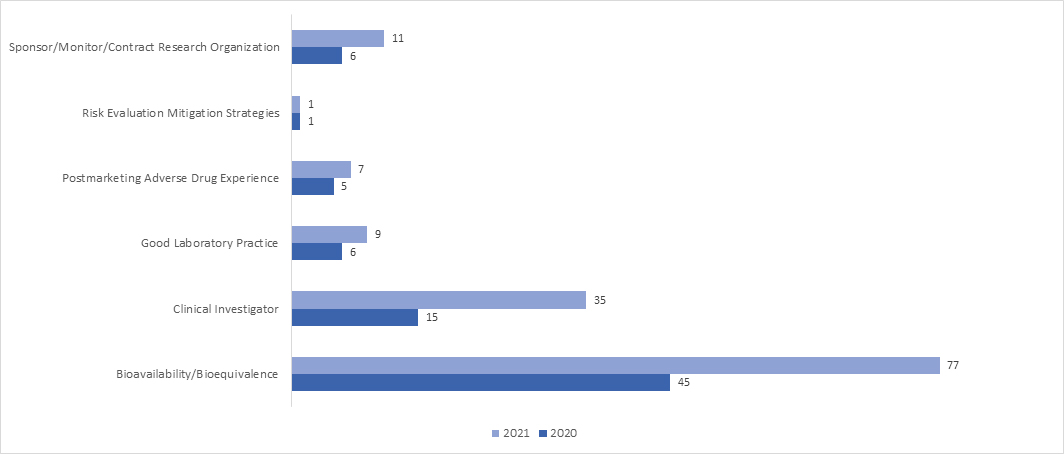
How many Remote Regulatory Assessments (RRAs) were conducted during the COVID-19 Pandemic under the BIMO Program?
In 2021, the adoption of RRA inspection significantly upsurged across all the programs. In April 2021, the FDA released a guidance document on “Remote Interactive Evaluations of Drug Manufacturing and Bioresearch Monitoring Facilities During the COVID-19 Public Health Emergency Guidance for Industry,” which provides exhaustive information on the FDA’s process of conducting RRAs.
What are the possible outcomes of a BIMO Audit?
During the BIMO audit, the US FDA may decide to take any of the below-listed actions based on compliance –
1. No Action Indicated (NAI)
NAI is applicable when the FDA field inspector has not identified any objectionable practice or only minor issues for which further action is not justifiable.
2. Voluntary Action Indicated (VAI)
VAI is applicable when objectionable practices have been identified but are not significant.
3. Official Action Indicated (OAI)
OAI is applicable when identified objectionable practices that compromise data integrity and/or human subject rights.
What are the most common non-conformances issued under BIMO Audit?
Some of the most common non-conformances observed during the BIMO Audit are –
- Not keeping a proper track of records
- Failure with respect to the investigational plan
- Failure to comply with the regulations
- Failure in monitoring the protocols
- Inadequate subject protection
- Inadequate accountability of the product that is being investigated
The BIMO audit is crucial for any developer or manufacturer of novel medical devices and technologies planning to launch their device in the US market. Complying with the regulations and guidelines to avoid any of the described pitfalls is quite important.
Do you need any assistance with respect to BIMO audit inspections? Reach out to Freyr. Stay informed. Stay compliant.