4 min read
Medical Device Reporting (MDR) is a post-market surveillance tools that the Food and Drug Administration (FDA) uses to monitor device performance, detect potential device-related safety issues, and contribute to benefit-risk assessments of devices. The purpose of MDR is to detect and address device related adverse events in a timely manner. It enables physicians, healthcare setups, manufacturers, and consumers for voluntary reporting to understand the device’s post-market safety and efficacy.
MDR is applicable to all classes of medical devices, which are either manufactured in the United States of America (USA) or imported to the USA. Medical Device manufacturers willing to market their devices in the USA must comply with MDR, otherwise which may lead to financial penalties. It is applicable in the USA including a foreign event, i.e. it is applicable to legally marketed medical devices in the United States both manufactured in the USA and foreign countries as well. Additionally, there are various cases of applicability for an MDR, such as:
- if a device is manufactured in the USA, distributed locally and to other markets
- when a device is manufactured in the USA but distributed in other markets
- when a device is manufactured in the foreign country, supplied in the USA and other markets
- when a device is manufactured in the foreign country and distributed locally and
- when a device is under investigation in the USA
MDR and the Reporting Process Flow
The MDR regulation contains many mandatory requirements for manufacturers, importers, and device user facilities to report certain device-related adverse events and product problems to the FDA. The below provided process flowchart details the reporting process step-by-step.
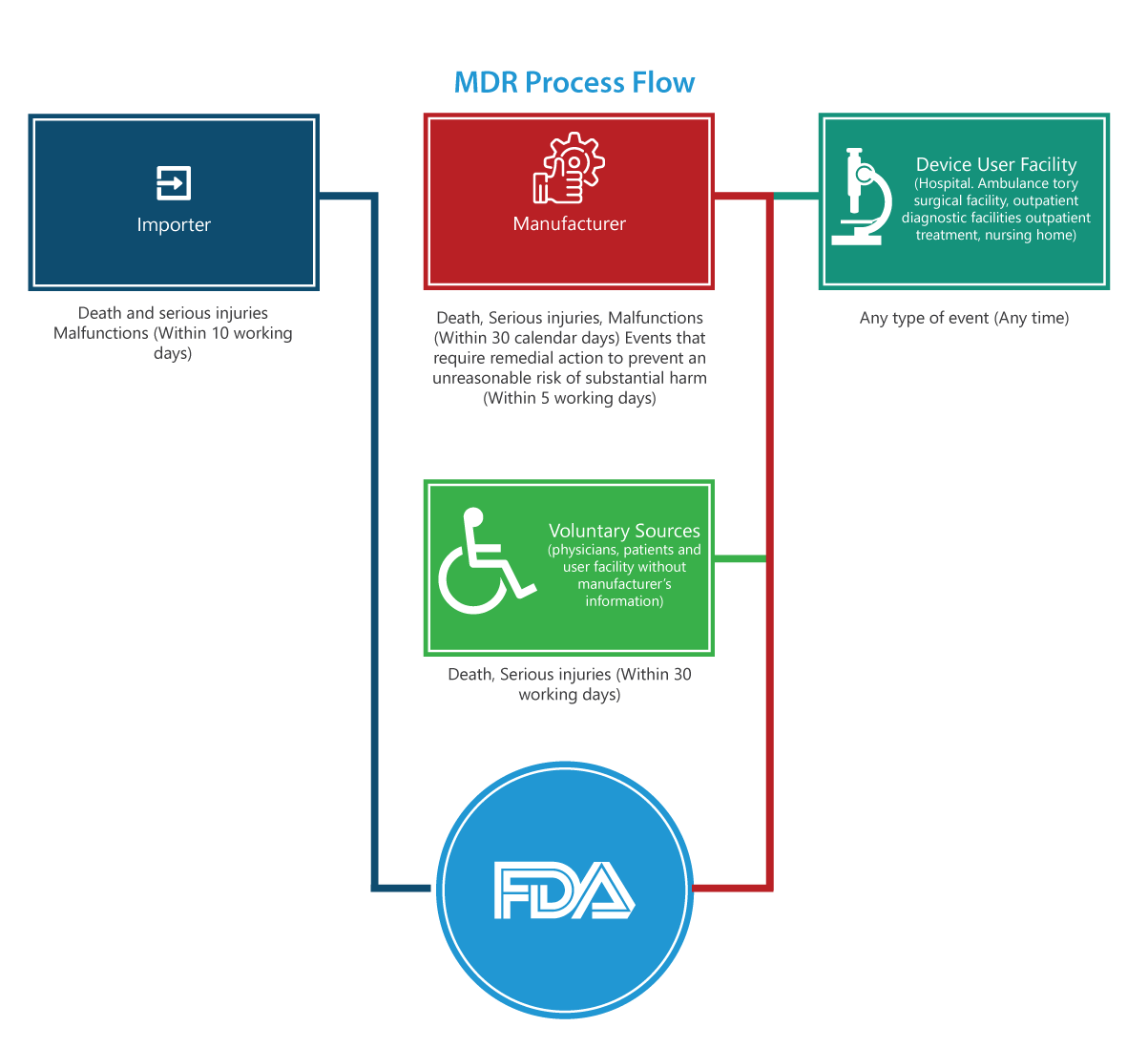
To whom does it apply?
Importers
Reports of deaths, serious injuries and malfunctions must be submitted to the FDA and the manufacturer within 30 working days. If the malfunction can cause injuries or deaths elsewhere, the importers are to report malfunction to the manufacturer.
Manufacturers
Reports for an event (deaths, serious injuries and malfunctions) designated by FDA or an event that requires remedial action to prevent an unreasonable risk of substantial harm to the public health must be submitted to the FDA within 5 working days by completing the 3500A form.
Device User Facility (Hospital, ambulatory surgical facility, nursing home, outpatient diagnostic facility, or outpatient treatment facility)
Reports must be submitted to the manufacturer of the device no later than 10 work days after the day that it becomes aware of information that a device has or may have caused or contributed to a serious injury to a patient of the facility. If the manufacturer is unknown, the facility must submit the report to the FDA.
Voluntary Groups
Patients, healthcare professionals and consumers who find a problem related to a medical device can report to FDA through MedWatch
eMDR
The FDA mandated electronic MDR (eMDR) in 2015 to identify critical issues of data quality and integrity associated with reporting serious injuries related to all classes of medical devices. eMDR is preferred to be the mode of reporting.
Manufacturers can submit their eMDR through an Electronic Submissions Gateway (ESG). From the time of submission, the electronic gateway takes up to 48 hours to send an acknowledgment. If there is any error while submitting report ‘a message’ will show up for making the correction(s).
eMDR – How is it beneficial?
eMDR offers multiple advantages over manual reporting mechanism (i.e., MDR). A few notable benefits that manufacturers / agency / patients can bank on are listed below:
- eMDR submission tool enhances better collaboration between an organization, the health agency (FDA), and patients.
- eMDR saves costs. Automation reduces the need for administrative overhead and traditional communication; it helps to speed up the process and fosters effective event reporting, resulting in immediate interaction with the FDA.
- Manual processes involve substantial paperwork, may be lengthy and difficult to track and process. eMDR submission is automated and centralized. Records can be retrieved easily, saving a lot of time while reviewing.
- eMDR enables parties to flag submission errors quickly, as opposed to manual and time-consuming correspondences with the FDA.
- eMDR acts as a single point of entry to process all electronic submissions in a highly secured environment and it is beneficial because complaints at the organization can be linked directly to the MedWatch form and integrated within the FDA’s gateway.
eMDR and the Reporting Process Flow
The eMDR regulation contains mandatory requirements for manufacturers, importers, and device user facilities to report certain device-related adverse events and product problems to the FDA. The flowchart below details the reporting process step-by-step.
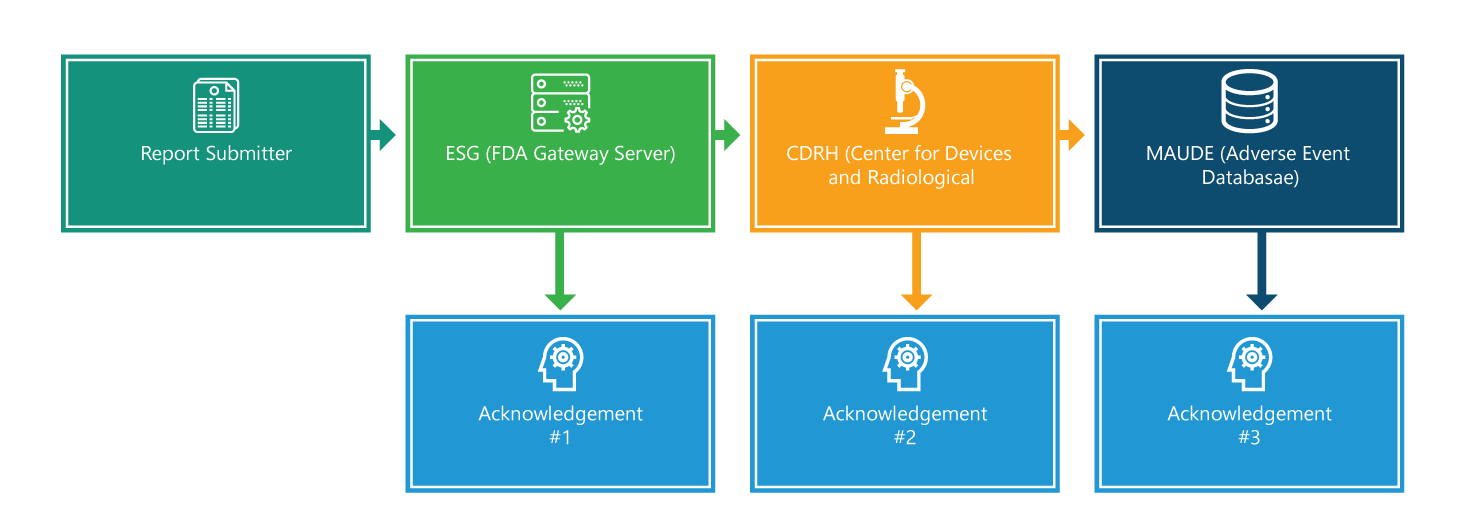
The reporting process comprises of four steps. Except the first step, each step is acknowledged. Further, each step is provided with additional information that will help ease the process.
Step 1: Report Submitter
Submitting an eMDR. At the outset, to make a submission, one should have an electronic signature and should ensure submission filenames include only one period, which is used to indicate the file type extension (for example 555.xml or 555.pdf). However, the application delivery and processing time depend on the overall size of your submission; larger submissions take longer time to get delivered and processed.
Step 2: Electronic Submissions Gateway (ESG)
When your submission reaches the ESG, you should quickly receive an acknowledgment #1 unless the ESG is down for maintenance. You are required to check the status of your MDR on the ESG web site.
Step 3: CRDH
eMDR is automatically routed from the ESG to the Center for Devices and Radiological Health (CDRH). Once it is routed, like in the step 2, you should receive an acknowledgment, i.e., #2.
Step 4: Manufacturer and User Facility Device Experience (MAUDE)
When the CDRH validates and updates the submission in the Adverse Event database (MAUDE), it is expected the submitter should receive an acknowledgment #3. It is to be noted that any errors that occur during the validation and loading are recorded.
Medical Device Reporting (MDR) is a critical process that helps save lives and protect patients from unnecessary risks. It ensures that all parties involved in patient care are responsible and alert in using devices.
eMDR makes reporting easier, but the documentation and follow up can be resource consuming. Get it right first time; speak to us at sales@freyrsolutions.com.