2 min read
US FDA’s focal motive is to constantly scrutinize and bridge the gap between the Regulatory processes for uninterrupted import and sale of new and high-quality medical devices in the US market. The US FDA in 1998 released a program called “The New 510(k) Paradigm: Alternate Approaches to Demonstrating Substantial Equivalence in Premarket Notifications.” It intends to establish an efficient FDA 510(k) submission pathway that contains certain changes in the already approved 510(k) application. This new 510(k) notification offers (03) three types of submissions, namely, special 510(k), abbreviated 510(k), and traditional 510(k). The US FDA, in 2019, issued a special 510(k) guidance document describing an optional pathway for manufacturers that make certain well-defined modifications to their legally marketed device.
Why a Special 510(k)?
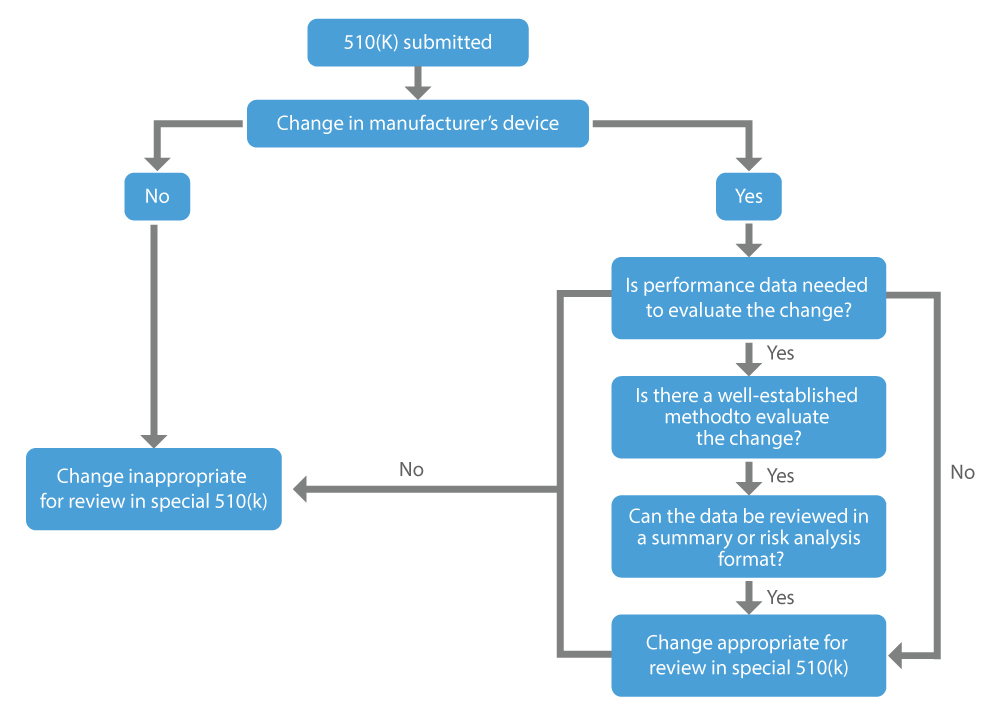
When a manufacturer wants approval for the modifications they have made in the already marketed device, i.e., the existing device, they can apply for a special 510(k). The main factors to consider when determining whether a change to an existing device may be appropriate for a special 510(k) are the following:
- The change is in the submitter’s own legally marketed predicate device.
- Performance data are not required, or well-established methods are available if deemed necessary to evaluate the change.
- All performance data to support a substantial equivalence determination can be reviewed in a summary or risk analysis format.
Documents Required for Special 510(k)
- Cover letter
- The name of the manufacturer’s legally marketed (existing) device and the 510(k) number
- A detailed description of the change(s) made to the device that resulted in the submission of a new 510(k)
- A comparison of the modified device to the cleared device in a tabular format
- Other changes to labeling or design
- A concise summary of the design control activities
- Based on the risk analysis, an identification of the verification and/or validation activities required to comply with 21 CFR 820.30
- Indications for use form
- A statement that the submitter has complied and is not currently in violation of the design control procedure requirements as specified in 21 CFR 820.30 and the records are available for review upon request
Special 510(k) Review Timeline by the US FDA
According to the FDA’s guidelines “Refuse to Accept Policy for 510(k)s”, the review timeline for special 510(k) submissions is within thirty (30) days of its receipt.
When to Apply for Special 510(k)?
The US FDA makes consistent efforts to provide safe and effective medical devices to promote human health. The Special 510(k) program is efficient and consistent with the least burdensome review procedure that helps foreign manufacturers to sell their devices within the USA and allows patients to get timely access to new medical devices.
For any further clarification on FDA’s special 510(k) process, reach out to Freyr - a proven Regulatory expert. Stay informed. Stay compliant.