

2 min read
The US FDA has published a guidance document that will help the industry and the Health Agency (HA) staff determine when a software change to a medical device requires a manufacturer to submit and obtain the FDA clearance of a new premarket notification (510(k)). This guidance intends to enhance the predictability, consistency, and transparency of the “when to submit” decision-making process by providing a least burdensome approach and describing the Regulatory framework, policies and practices underlying such a decision, specifically related to software changes. Let’s uncover the FDA guidance in detail.
FDA Guiding Principles and Flowchart
With an intent to assist the medical device manufacturers in applying the main principles, the document provides a flowchart, additional clarifications and examples needed to make decisions for a new premarket notification 510(k) for a software change made to an already approved device in the US. Additionally, one should follow several guiding principles while using this guidance to determine whether to submit a new 510(k) to change an existing device. Some of them are widely known and derived from the current FDA 510(k) policy and the others are necessary to use the logic scheme mentioned in this guidance. As per the guidance, the scheme provided cannot cover all the possible intricacies related to such changes and how they affect the decision. Therefore, to determine the necessity of a new premarket notification 510(k), medical device manufacturers should consider the general principles and the flowchart as summarized below.
- Changes that affect the safety or effectiveness of a device
- Initial risk-based assessment
- Unintended consequences of changes
- Use of risk management
- Role of testing (verification and validation activities) in evaluating whether a change could significantly affect safety and effectiveness
- Evaluating simultaneous changes to determine whether the submission of a new 510(k) is required
- Appropriate comparative device and the cumulative effect of changes
- Document requirement (21 CFR Part 820)
- 510(k) submissions for modified devices
- Substantial equivalence determinations
- Submission of a new 510(k) is likely required if a manufacturer modifies their device to affect the safety or effectiveness of the device. However, changes that are not intended to affect a device’s safety or effectiveness should still be evaluated.
- To determine whether a change or modification could significantly affect the safety or effectiveness, the manufacturer should first conduct a risk-based assessment to know whether the change could affect the device’s safety or effectiveness either positively or negatively. This risk-based assessment should identify and analyze all new risks and changes in existing risks resulting from the device change and lead to an initial decision on whether the submission of a new 510(k) is required.
- Sometimes there are additional unintended or unplanned consequences that may be triggered during software submissions. The flow chart should assess these consequences to determine if the submission of a new 510(k) is needed.
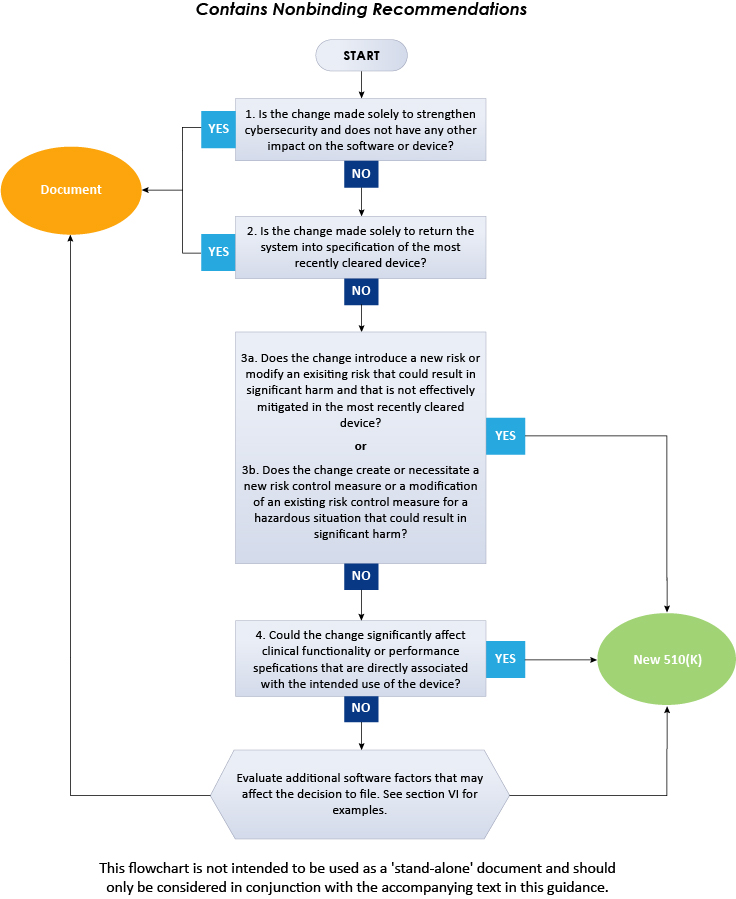
The above flowchart illustrates a step-by-step procedure to be followed to decide on 510(k) submission for software changes in existing devices. In conclusion, the present FDA guidance describes in detail the approach to be followed by the medical device manufacturers when deciding whether the software changes to an existing medical device require submission of a new 510(k). To gain further insights on the FDA guidance, consult Freyr - a proven Regulatory expert. Stay informed. Stay compliant.









