

3 min read
510(k) is a premarket submission made to the FDA to demonstrate that the device to be marketed is as safe and effective, that is, substantially equivalent to a legally marketed device (predicate). Devices with moderate risk are required to submit a 510(k) notification, which includes a minority of Class I and III devices and a majority of Class II devices.
There are three (03) types of 510(k) programs, Traditional, Abbreviated, and Special. The safety and performance pathway was introduced in 2019 and built on the abbreviated program. The eSTAR program introduced in 2020 allows for a comprehensive medical device submission through an interactive PDF form.
Who Needs a 510(k) Certification?
510(k) is essentially the name of the process/pathway that medical device manufacturers intending to market their moderate to high-risk devices in the US undergo to demonstrate that the product to be marketed is as safe and effective as a legally marketed device.
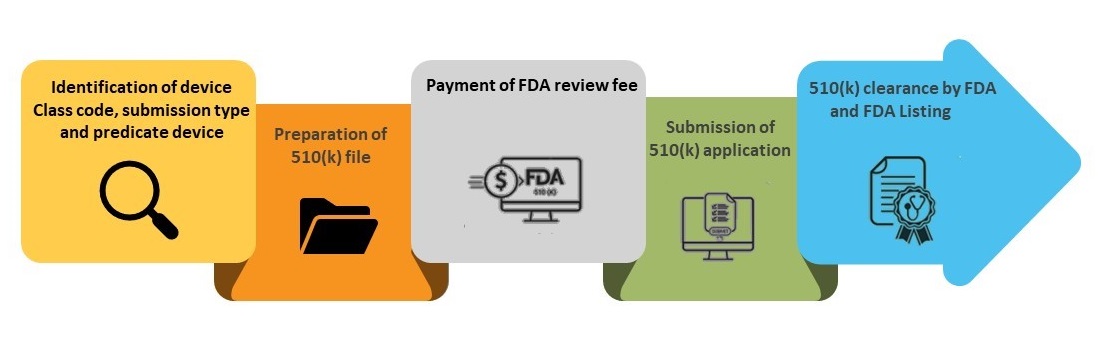
Detailed below is the step-by-step process for obtaining a 510(k) clearance.
Step 1- Identification of Device Class Code, Submission type, and Predicate Device
- Identify product code and regulation number- To determine the 510(k) test requirements, it is necessary first to identify the product code and regulation number. One can start a search on the FDA database to find the 7-digit regulation number for which the identification matches the intended use of the device in question.
- The FDA product code consists of three (03) letters. Information regarding product classification, regulation description, and GMP requirements can be located using this code.
- Selection of submission type- A submitter may choose one of the three (03) submission types mentioned earlier. Traditional 510(k) is for first-time submissions, Special 510(k) is for medical device manufacturers who want to submit changes to an existing device and Abbreviated 510(k) can be chosen when the device complies with established voluntary consensus standards. In case of an Abbreviated 510(k), the submitter must rely on FDA guidance documents.
- Identification of predicate device- A medical device manufacturer needs to prove that the device they intend to market has the same intended use and technical characteristics as the legally marketed device, also known as the predicate device. If there are any differences in technical characteristics, the submitter must prove that no safety and effectiveness concerns are associated with the difference.
Step 2- Preparation of 510(k) File
The next step is to prepare the 510(k) file, the guidance, and the information, which is available on the FDA website. It includes the acceptance checklist for all three (03) types of 510(k) programs and a microsite titled Content for 510(k), which includes information regarding statements for indications for use, substantial equivalence comparison, and proposed labeling, among other useful information.
510(k) submission process steps
Step 3-Payment of FDA Review Fee
All types of 510(k) applications are subject to the user fee. For the financial year 2023, the standard fee for 510(k) is $19,870. For businesses certified under the Centre for Diagnostics and Radiological Health (CDRH), also known as small businesses, the fee is $4,967. The fee is subject to change in the next financial year.
Step 4- Submission of 510(k) Application
The submitter can send an electronic copy (eCopy) or an electronic Submission Template and Resource (eSTAR) premarket submission through the CDRH portal.
Starting October 01, 2023, all 510(k) submissions, unless exempted according to the final guidance, must be submitted as electronic submissions using eSTAR.
After the 510(k) is submitted, a unique control number is assigned, which is known as the “510(k) number” or “K number.” FDA conducts two verification checks, one to verify if the proper user fee has been paid and the second to verify if a valid eCopy or eSTAR was provided.
- By day 07, FDA sends an acknowledgment letter in case the proper user fee is paid and a valid eCopy or eSTAR is provided. If not, the FDA sends a hold letter for unresolved issues.
- By day 15, FDA conducts an acceptance review FDA informs the submitter if 510(k) is accepted for substantiative review or placed on Refuse to Accept (RTA) hold.
- By day 60, FDA conducts a substantiative review. FDA communicates via
substantiative interaction to inform the FDA will either proceed with an interactive review, or the 510(k) will be placed on hold, and Additional Information will be requested.
Step 5- FDA Clearance and Listing in FDA 510(k) Database
The goal of the FDA is to announce its Medical Device User Fee Amendments (MDUFA) decision in 90 FDA days. FDA days are the calendar days between the date the 510(k) was received and the date of an MDUFA decision, excluding the days the submission was on hold for an additional information request. MDUFA decisions for 510(k) submissions include findings of substantially equivalent (SE) or not substantially equivalent (NSE).
When a decision is made, FDA issues a decision letter to the submitter by email. A 510(k) application that receives an SE decision letter is deemed “cleared.” It is then listed in the 510(k) database along with Indications for the use of the medical device and the 510(k) summary or 510(k) statement as attachments.
It can be concluded that careful planning and execution through thorough documentation and an in-depth understanding of the Regulatory environment are crucial for a successful 510(k) submission to FDA.
For assistance with the 510(k) submission process of your medical device, you can write to us at sales@freyrsoltions.com, or schedule a call with our experts, who can help you navigate the procedures. Stay informed. Stay compliant.









