

5 min read
Medical device software in South Korea is used to diagnose, treat, and monitor patients in the modern healthcare system. It comprises both embedded software that is integrated into medical devices and standalone software that may be utilized on PCs, mobile devices, and web-based services. The Ministry of Food and Drug Safety (MFDS), South Korea is responsible for regulating medical device software and ensuring its safety and efficacy. On July 05, 2023, the MFDS laid out criteria for the approval and inspection of medical device software; these regulations provide a structure that civil petitioners can follow when submitting software for approval or review.
The regulations address a variety of subjects, including the scope of the application, technical documentation requirements, and compliance verification reports. There are international standards and guidelines that apply to medical device software, in addition to the MFDS guidelines, such as the International Electrotechnical Commission (IEC) 62304 standard for software life cycle processes and the United States Food and Drug Administration (US FDA) guidance on mobile medical applications.
Software Development Plan and Requirements Analysis
- The Software Development Plan outlines the overall approach to software development, including specifications, methods, and development tools. It also covers verification, medical device risk management, configuration management, and documentation.
- Requirements Analysis establishes the medical device software requirements, including risk control measures and verification methods. By carefully planning and analyzing the software development process, developers can ensure that the resulting software meets the necessary standards of safety and efficacy.
- The Software Conformity Verification Report includes an outline of the software development plan, the manufacturer’s document control number, and an overview of the requirements analysis. By adhering to these guidelines, medical device software can be developed with confidence, knowing that it has undergone rigorous testing and meets the necessary standards of safety and efficacy.
Medical Device Software Verification and Validation
- Medical Device Software Verification ensures that the software meets the specified requirements.
- Medical Device Software Validation ensures that the software meets the user’s needs and the intended use(s).
- Medical Device Software Verification and Validation Report outlines the verification and validation process, including the product name, revision and the names of the individuals who examined and approved the report. The report may vary, depending on the characteristics of the software, but it should include a description of the software, the verification and validation methods used, and the results of the testing.
Operating Environment and Software of Unknown Provenance (SOUP)
- If the software is dependent on specific hardware, such as embedded software, the technical document should describe the hardware specifications.
- However, if the software is standalone and developed to work on general-purpose hardware, the operating environment must be described in the raw material. This includes the minimum recommended specifications, such as Microsoft Windows 10 or above.
- Additionally, if the medical device software includes commercial Software of Unknown Provenance (SOUP), an operating environment must be created to ensure proper functioning. By carefully describing the operating environment and addressing any SOUP, developers can ensure that their medical device software is safe and effective for its intended use.
Medical Device Risk Management and Documentation Requirements
- The software as a medical device risk management process includes identifying hazardous situations, establishing risk control measures, verifying those measures, and managing software changes.
- The MFDS-RM software risk management document provides information on software risk management.
- Additionally, documentation requirements are essential to ensure that the software meets the necessary standards of safety and efficacy.
- The software development plan, medical device software requirements analysis, and software verification and validation reports must be included in the documentation.
- The Software Conformity Verification Report outlines the documentation requirements; it also includes an outline of the applicable documents and the manufacturer’s document control number.
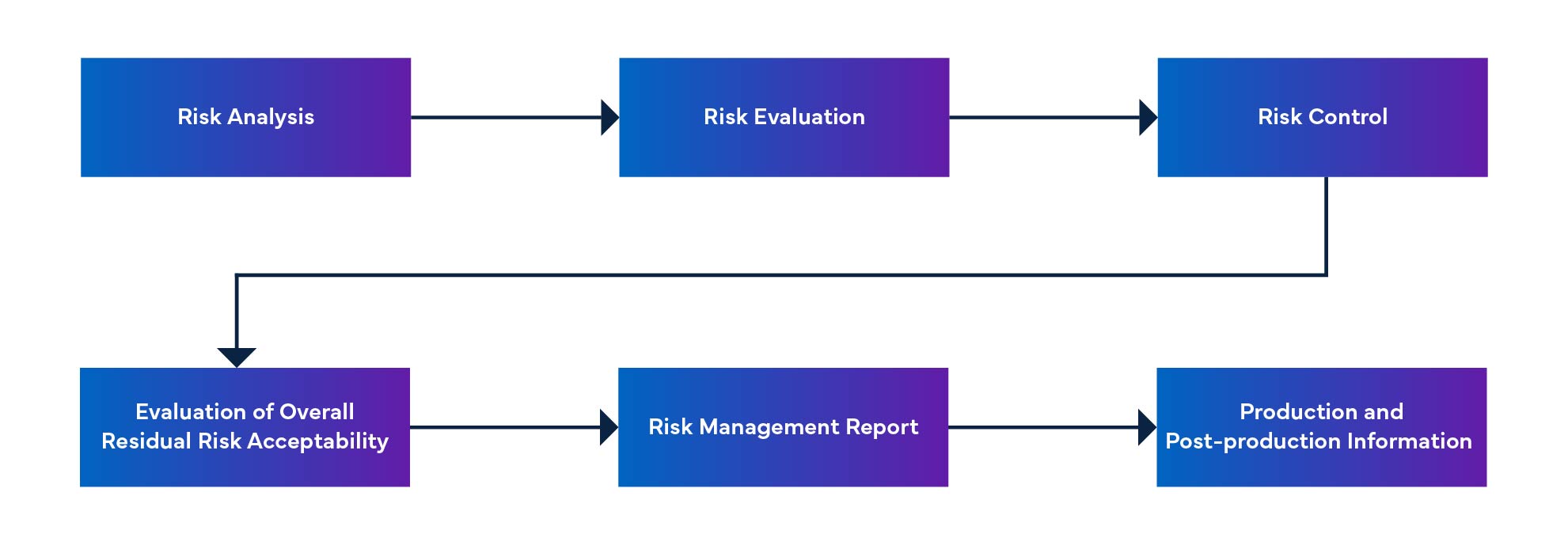
Figure 1: Medical Device Risk Management Process
Unresolved Anomalies and Corrective Actions for SaMD Software
- The MFDS-PR (Software Problem Resolution) document outlines the software problem resolution process, which includes problem reporting, analysis, implementation, and verification.
- The document also includes a list of unresolved problems, bugs, defects, and anomalies, as well as a residual risk assessment for the software system.
- The corrective actions taken to address these issues must be documented in the software maintenance plan, which is established according to the software maintenance process.
- The MFDS-maintenance document provides information on SaMD software maintenance and troubleshooting.
Technical Document Review and Submission Requirements for SaMD Software
The main review documents during the review process are performance data, conformity confirmation report, and medical device software verification and validation data, the Software Design Specification (SDS), medical device Software Requirements Statement (SRS), and verification and validation reports. The Conformity Confirmation Report and Medical Device Software Verification and Validation Report must be submitted.
Medical Device Software Risk Management
- Identifying potential hazards associated with the software and its use.
- Assessing the severity of risks associated with these hazards.
- Implementing risk control measures to minimize the likelihood of harm.
- Monitoring and reviewing the efficacy of these risk control measures.
- Documenting all medical device risk management activities and decisions.
In a software system, software items are broken down into smaller parts, including detailed software items. When an item cannot be broken down any further, it is called a unit. The system allows breaking down to the unit level, helping determine the safety level for each software item. By putting these software items together, we are able to figure out the safety level for the entire software system.
Figure 2: Medical Device Software Disassembly and Integration
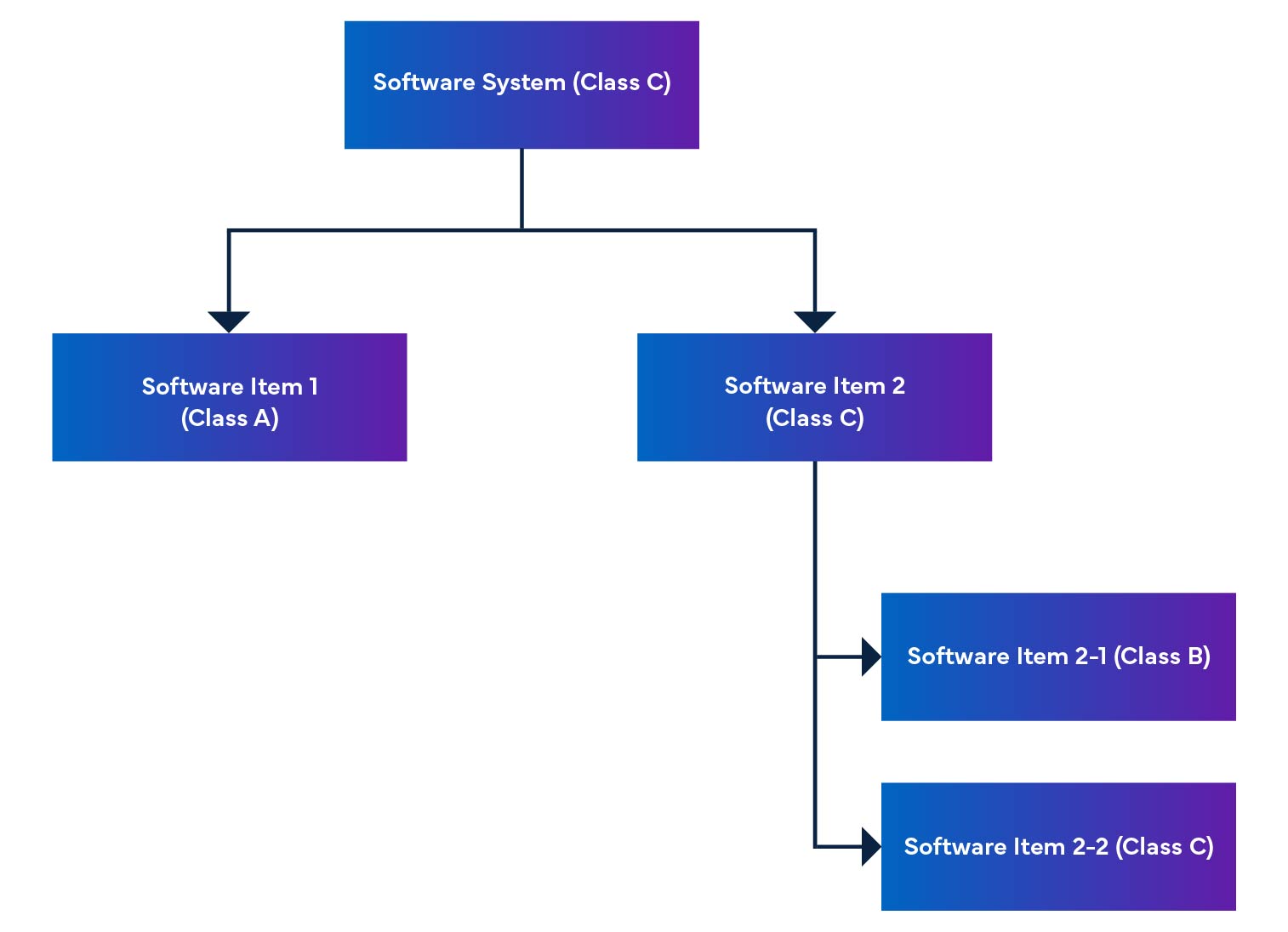
The regulation also mentions software safety rating, which is a rating for identifying the risks of SaMD software (see Table 1).
Table 1: Definition of Safety Rating
| Rating | Medical Device Software Safety Class Definition |
| Class A | No possibility of injury or bodily harm. |
| Class B | Less serious injuries (minor injuries) are likely. |
| Class C | Possibility of serious injury or death. |
Software Configuration Management
- Maintaining accurate and up-to-date documentation for all software versions, changes, and updates.
- Ensuring that all documentation is properly reviewed and approved.
- Implementing procedures for managing software configuration changes.
- Documenting all software configuration management activities and decisions.
Software Maintenance
- Regularly testing and monitoring the software to ensure that it remains safe and effective for its intended use.
- Implementing procedures for addressing any issues that may arise, including bug fixes and software updates.
- Documenting all software maintenance activities and decisions.
Troubleshooting
- Identifying the root cause of the issue.
- Implementing corrective actions to address the issue.
- Documenting the entire troubleshooting process for future reference.
By following the above guidelines, developers can ensure that any issues with their medical device software are properly addressed and documented, and that the software meets the necessary requirements for approval or examination.
If you are a medical device manufacturer aiming for compliance with South Korean medical device software standards, Freyr’s Regulatory experts can guide you through the intricate Regulatory landscape of the country. We will ensure that your devices align with the latest South Korea medical device regulations for seamless compliance. Contact us to learn more!









