Welcome to the May 2015 issue of FREYRFOREWORD!
A monthly round-up of the latest happenings and updates from Freyr.
Happy Reading!
CLINICAL TRIAL DISCLOSURE: EVER-CHANGING LANDSCAPE AND IMPACT
 Clinical trials are research studies that explore whether a medical strategy, treatment or device is safe and effective for humans.
Clinical trials are research studies that explore whether a medical strategy, treatment or device is safe and effective for humans.
IN PERSPECTIVE: REGULATORY AFFAIRS CONSULTING
 The Life sciences industry is undergoing a plethora of dynamic changes on a global scale.
The Life sciences industry is undergoing a plethora of dynamic changes on a global scale.
CELEBRATING NEW CLIENT WINS
 Our latest new client wins are a fantastic fit for our regulatory expertise and services, we’re absolutely delighted to be working on these projects for one of the Top Global Pharmaceutical Companies.
Our latest new client wins are a fantastic fit for our regulatory expertise and services, we’re absolutely delighted to be working on these projects for one of the Top Global Pharmaceutical Companies.
FREYR SUCCESS STORY
 Learn how Freyr established and streamlined master dossier management process for a Top 3, Fortune 50, $40+ Billion Pharma / Consumer Company.
Learn how Freyr established and streamlined master dossier management process for a Top 3, Fortune 50, $40+ Billion Pharma / Consumer Company.
FREYR SUCCESS STORY
 Learn how Freyr provided strategic clinical report publishing services for a National Biotech and Medical Research Agency.
Learn how Freyr provided strategic clinical report publishing services for a National Biotech and Medical Research Agency.
 |
||
| INTRODUCTION Clinical trials are research studies that explore whether a medical strategy, treatment or device is safe and effective for humans. These studies also may show which medical approaches work best for certain illnesses or groups of people.
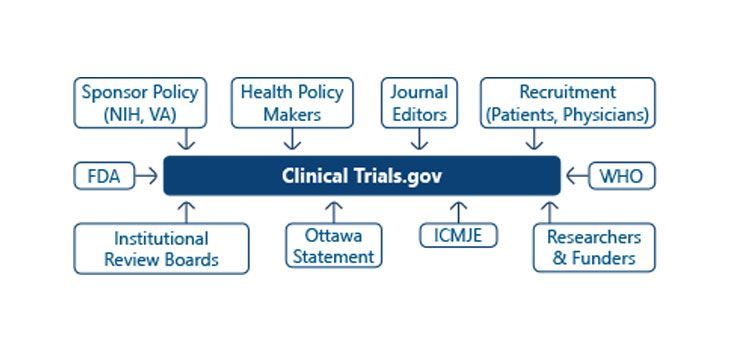
IMPORTANCE OF CLINICAL TRIAL DISCLOSURE: Life sciences organizations are enabled to efficiently manage clinical trial registration and reporting of results to clinicaltrials.gov, EudraCT and other registries.It offers support to the entire process of gathering data by stakeholders for review and conducting checks against registry specific validation rules and generating XML files for upload to the registry. It will also help companies to maintain compliance and consistency of data published in registries worldwide. CLINICALTRIALS.GOV: ClinicalTrials.gov contains information about medical studies in human volunteers.This web site and database of clinical studies is commonly referred to as a “registry” and “results database.” ClinicalTrials.gov does not contain information about all the clinical studies conducted in the United States because not all studies are required by law to be registered.
The results database was made available to the public in September 2008. FDAAA 801 also established penalties for failing to register or submit the results of trials. In the US, a sponsor needs to submit the data for protocol results to CTGOV and independently goes to the FDA asking for approval to conduct the trial. RESULTS DATABASE:
RESULTS ENTRY AHEAD OF CLINICAL TRIAL: GENERAL RESULTS AHEAD OF CLINICAL TRIAL: Recruitment Details: Key information relevant to recruitment process for overall study, such as dates of recruitment period and types of location (e.g., medical clinic). Pre-assignment Details: Definition: Description of any significant events and approaches for overall study (e.g., wash out, run-in, transition) following participant enrolment, but prior to group assignment. Arm/Group: Arm/Group Title: Label used to identify the arm or comparison group. Arm/Group Description: Brief description of the arm or comparison group to distinguish it from other arms/groups in the trial. Period(s): Discrete stages of a clinical trial during which numbers of participants at specific significant events or points of time are reported (There is no limit to the number of periods). Period Title: Title describing a stage of the trial. Number of participants required at the beginning of the period. Baseline Characteristics: A table of demographic and baseline data (study specific) for the entire trial population and for each arm or comparison group. Results Described During/After Clinical Trial: Results of applicable clinical trials must be reported within 12 months following the primary completion date. Primary Completion Date is the date the final subject was examined or received an intervention for the purposes of final collection of data for the primary outcome, whether the clinical trial concluded according to the pre-specified protocol or was terminated. “Study Completion Date” is the final date on which data was collected. Regardless of the outcome, after completion or termination of a clinical trial in patients, the results of the trial are posted according to current laws and regulations. GENERAL RESULTS DESCRIBED DURING TRIAL: Completed: Number of participants completed the study. Discontinuation details: Number of patients discontinued from the study. Reason Not Completed: Additional information about participants who did not complete the period, if any. Demographics of participants: (Ex., age, sex, race, etc.) Laboratory results: Haematology, biochemistry and serological results of all the participants. Safety parameters: Safety parameters include laboratory and other vital check-ups for all the participants. Efficacy parameters: Efficacy details of the IMP and significant changes with the comparator drug. Comments: Additional information about the completed milestone. Adverse Events: Two types of adverse event data are to be reported
Details of Serious Adverse Event (per arm/group): Overall number of participants affected by one or more Serious Adverse Events. Overall Limitations and Caveats: If appropriate, describe significant limitations of the trial. CLINICAL TRIAL REGISTRATION PROCESS:
Content Courtesy: ClinicalTrials.gov |
 |
||||
| ARE YOU DERIVING VALUE OUT OF THE ENGAGEMENT? The Life sciences industry is undergoing a plethora of dynamic changes on a global scale. With globalization opening immense potential for huge business gains across rapidly growing markets, the strategic move to manufacture, market and operate in global economies is increasingly meeting the complex challenges of stringent regulatory adherence requirements.In such a scenario, companies are increasingly looking to engage with the right Consulting Partner who can help them successfully navigate the regulatory challenges.The aim is to achieve optimized Regulatory and Compliance procedures with minimal risks through an optimal cost effective option.The Regulatory Affairs consultation provides necessary assistance during the pharmaceutical development process, which in turn helps companies in the implementation of a Global Regulatory Strategy.
The major concern is how well a consultant analyses the Regulatory Compliance obligations and values the company’s needs.Here are some of the common parameters that, in principle, help a company measure the credibility and derive value out of its consultative engagement. |
||||
| RIGHT PARTNER VS RIGHT VALUE Identifying the right consultant, possessing the expertise, knowhow and experience, is the most critical aspect. Solution partners with industry expertise can leverage their consultative and solution implementing experience to ensure a company streamlines the regulatory processes to meet the objectives, within mandated timelines and also saves significantly on cost of compliance.They also impart knowledge regarding the current standards and latest rules of regulatory agencies, analyze the gaps and the needs to meet them and provide the action points to publish the submissions within the stipulated time.Look out for some of the few challenge areas mentioned below that a Consultant should address effectively:
There are other critical areas that will demand your attention during a compliance life cycle. And, if you find yourself asking all the questions but getting no definitive solution roadmap, it’s time to look for a partner who brings the experience, the expertise and, more importantly, who brings value to you! GLOBAL REGULATORY AFFAIRS CONSULTING MARKET OVERVIEW
In 2013, the regulatory writing and publishing services segment reportedly took 40% share out of the total regulatory affairs outsourcing market owing to growing need for rapid approval and submission of drug applications. In addition, the understanding of global and domestic regulatory requirements by regulatory writers helps in driving the growth of the regulatory writing and publishing services market. It is reported that during 2014 to 2020, the regulatory consulting and legal representation services segment is estimated to have the fastest growth rate in the regulatory affairs outsourcing market. This is owing to growing pressure for regulatory consulting and increasing need of client interaction with various regulatory agencies. The global regulatory affairs outsourcing market is segmented by services into regulatory affairs; clinical trial applications and product registrations; regulatory writing and publishing; regulatory consulting and legal representation; and others (post approval maintenance, reimbursement consulting etc.). In 2013, the regulatory writing and publishing services segment reportedly took 40% share out of the total regulatory affairs outsourcing market and is expected to maintain its lead during the forecast period through 2014 to 2020. Increasing need for user/reviewer friendly and complete drug application submissions and sound knowledge for drug development services are some of the factors which have contributed towards the large market size of the regulatory writing and publishing services. Furthermore a well-written clinical study report (CSR) adds a lot of value in the final production of clinical trial documentation, which is in compliance with stringent regulatory requirements of the drug development process. It is also stated that the regulatory consulting and legal representation services segment is expected to increase at a compound annual growth rate (CAGR) of above 14% owing to growing demand of drug manufacturing companies for product safety and efficacy evaluations. In addition, growth of the regulatory consulting and legal representation services segment can be increased if pharmaceutical companies try and minimize business impacts such as product recalls and loss of sales ensuring global environmental compliance. In 2013, North America took the largest market share for the global regulatory affairs outsourcing market owing to increased number of clinical trial activities, vast presence of research units and cost benefits incurred by shifting high costs of in-house resources for various regulatory activities. Asia Pacific is predicted to have the highest growth rate over the forecast period that is attributed to availability of large population base and skilled workforce. Over the next few years, there is scope of increased contract manufacturing activities in emerging nations such as China, India, Malaysia and Vietnam which will give rise to growth of the global regulatory affairs outsourcing market in Asia Pacific region. |
||||
| IN CONCLUSION: The FDA intends to enable product development and manufacturing flexibility and supports advancement of innovative ways to lower the cost of drug development while advancing the critical path for drug products. It is likely that third-party outsourcing service providers will play a pivotal role in delivery of business functions like drug development, sales and marketing and regulatory compliance services. Pharmaceutical companies must strategize the execution of services they need to outsource and select a supplier/partner with good track record, in-depth industry knowledge and must possess excellent business relationships with regulatory agencies.The ideal service provider should also have the staff capacity to perform the developmental and registration activities. In 2013, the global regulatory affairs outsourcing market was valued at $1.56 billion stated a recent industry report. It is expected to grow at a CAGR of 14.6% from 2014 to 2020 to reach an estimated value of $4.49 billion.As Regulatory consultant and global solutions partner to several Top 10 Fortune companies, handling end-to-end multi-geo Regulatory Affairs responsibilities across their Top 20 global brands, Freyr is best positioned to provide proven expertise and tailored services that put you in total control of your entire global Regulatory compliance obligations. |

 |
||
| As an organization, we at Freyr, have always placed the highest value on our business associations and partnerships. It has been our guiding principle to identify newer opportunities and create exceptional engagement excellence for our clients that transform into long-term relationships. As always, it is a great pleasure to announce the New Wins. | ||
| STRATEGIC LABELING MANAGEMENT PROJECT FOR GLOBAL $1+BN, PHARMA COMPANY:
|
 |
||||||||||||||
| HIGHLIGHTS
BUSINESS IMPERATIVES 24 CMC Variations (amendments) for established medicinal product for treatment of hemorrhoids and other related anorectal conditions in 12 African countries. CHALLENGES
FREYR SOLUTIONS & SERVICES
CLIENT BENEFITS
|
||||||||||||||
 |
||||||||||
| HIGHLIGHTS
BUSINESS IMPERATIVES
CHALLENGES
FREYR SOLUTION & SERVICES
CLIENT BENEFITS
|
||||||||||