US FDA Medical Device Registration Overview
United States of America (USA) is renowned for being highly regulated market for Medical Devices with well-defined registration pathways and requirements. The initial medical device regulations of the U.S. date back to 1976 and have evolved over period. They are regulated by the Centre for Devices and Radiological Health (CDRH) under the Food and Drug Administration (FDA). Freyr has helped multiple device manufacturers to comply with the US FDA medical device registration process.
![]()
Regulatory Authority: Food and Drug Administration (FDA)![]()
Regulation: Title 21 Code of Federal Regulations (21 CFR) Parts 800 – 1299![]()
Regulatory Pathway: Pre-Market Notification or Pre-Market Approval or De-Novo Classification![]()
Authorized Representative: U.S. Agent![]()
QMS Requirement: Quality System Regulation (QSR) (21 CFR part 820)![]()
Assessment of Technical Data: Centre for Devices and Radiological Health![]()
Validity of License: Unlimited![]()
Labeling Requirements: 21 CFR Part 801![]()
Submission Format: Paper & CD/DVD![]()
Language: English
Medical Device Classification USA
FDA classifies Medical Devices into 3 risk-based categories, Class I, Class II and Class III wherein Class I devices are considered as low risk devices, and Class III are associated with high risk. The registration requirements and pathway vary with class of device.
| Device Class | Risk | Registration Pathway for approval |
|---|---|---|
| I | Low Risk | 510(k) exempt |
| II | Moderate Risk (With predicate device) | Premarket Notification/510(k) |
Moderate Risk (Without Predicate Device) | De-Novo application | |
| III | High Risk | Premarket Approval (PMA) |
U.S. FDA Agent
Companies with no local offices in the U.S. must appoint a U.S. FDA Agent to represent the manufacturer. The U.S. FDA agent must either reside in the U.S. or maintain a place of business in the U.S. The responsibilities to be fulfilled by the agent are pre-determined by the US FDA as a part of CFR regulations.
Navigate through the Frequently Asked Questions (FAQs) about US Agent.
Interactive Meetings with the US FDA
US FDA supports the manufacturers through various types of Q-Submission meetings to fulfil different objectives. Such meetings with the agency before initiation or during device development, prior to submission of US FDA medical device registration applications help manufacturers to optimize timelines and cost incurred for device commercialization.
Medical Device Registration USA
The devices can be approved by the CDRH, FDA through any of the various registration pathways. They are listed as:
Class I Medical Devices: The class I devices are usually GMP and 510(k) submission exempt and do not require prior approval from the US FDA to market them in the U.S. Other requirements such as Establishment registration, Device listing, UDI, PMS etc. must be complied by the manufacturer.
Class II Medical devices: Medium risk devices with 510(k) approved predicate devices can opt for 510(k) Pre-market Notification (PMN), also called 510(k) registration. The subject device shall establish Substantial Equivalence (SE) with the identified and claimed predicate devices. This pathway is most widely adapted pathway for registration of devices in the U.S. Manufacturers of medium risk devices with no predicates may request for classification by the US FDA through De-Novo applications.
Class III Medical Devices: High risk class III device manufacturers must submit a Pre-Market Approval (PMA) application to the US FDA. The devices must undergo detailed clinical evaluation and manufacturer must submit detailed safety and efficacy data from clinical studies. The US FDA would carry out QMS inspection as a part of assessment before issuing a Pre-Market Approval for the device.
Non-CDRH Medical Device Registrations
Based on indications of use, some border line products considered as medical devices in other countries such as, surgical respirators, disinfectants, combination products involve other Agencies such as, Centre for Disease Control (CDC), National Institute for Occupational Safety and Hazards (NIOSH), Environmental Protection Agency (EPA), Centre for Biological Evaluation and Research (CBER), Centre for Drug Evaluation and Research (CDER).
Post Approval Compliance Requirements for Medical Devices
All device manufacturers must comply with below listed post approval requirements:
- Registration and Listing Requirement: Establishments of all device classes must be registered with FURLs database and the device must be listed after the approval is obtained and prior to marketing of device in the U.S. Some devices such as, radiation devices must comply with other requirements such as, accession number before they can be imported into U.S.
- Unique Device Identification: All classes of devices must comply with the Unique Device Identification (UDI) regulations to market the devices in U.S.
- Establishment Fees: The manufacturer must pay the annual establishment fees to maintain their establishment registration active and to continue marketing devices in the U.S. The US FDA has reduced fee structure for smaller entities with active Small Business Certificate.
- Quality Audits: For devices which are not GMP exempt the US FDA may inspect the manufacturing establishment at any time for compliance with Quality Systems Regulations (QSR) in accordance with the 21 CFR 820.
Process flow
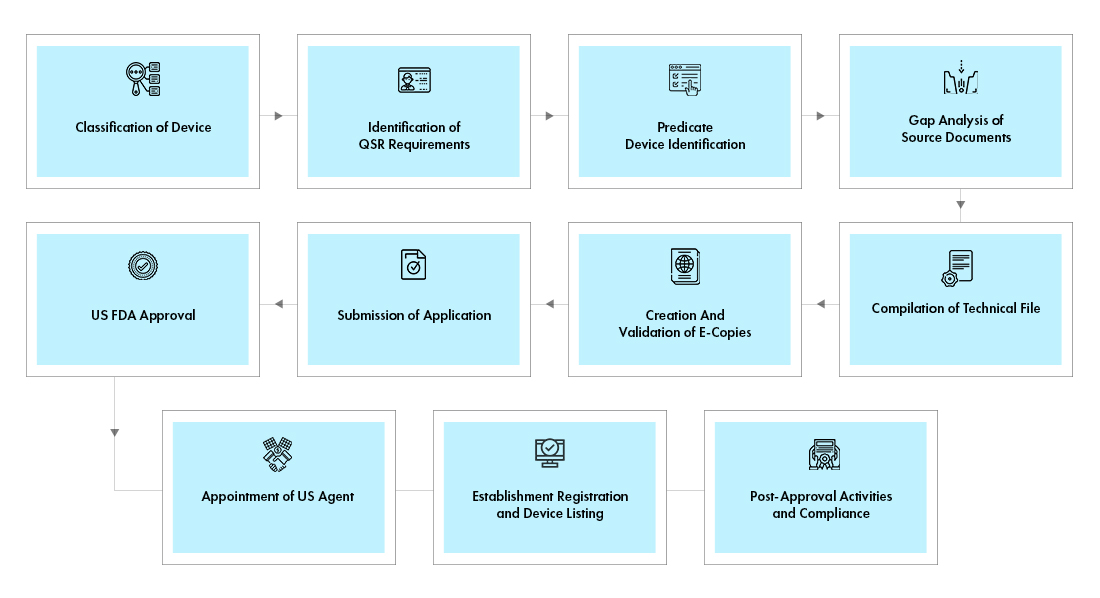
Post Approval Device Life Cycle Management
Freyr supports foreign manufacturers in end-to-end Medical Device lifecycle management, including post approval activities, such as:
- Post approval change management - modifications to existing medical device approvals such as, addition of new variants, accessories; addition of new indications of use among others
- Maintenance of approvals and registration through timely payment of MDUFA fees to the FDA
- Liaising between the US FDA and Manufacturer
Freyr has exclusive delivery center in the U.S. with professional team to provide Regulatory support for manufacturers in maintaining quality and safety needed for approval. Freyr’s intelligence experts keenly observe Regulatory updates and keep the clients informed about steps to be taken for product compliance with current standard.
Summary
| Risk | Device Class | QMS Audit | Predicate Availability | Regulatory Pathway | U.S. Agent | US FDA Timelines |
|---|---|---|---|---|---|---|
| Low Risk | I | No | NA | Exempted | Yes | 1 Month |
| Medium Risk | II | Yes (post approval) | Yes | PMN/510(k) | Yes | 9 - 12 Months |
| Medium Risk | II | Yes (post approval) | No | De-Novo Classification Request | Yes | 18 - 30 Months |
| High Risk | III | Yes (pre approval) | NA | PMA | Yes | 18 - 30 Months |
Freyr’s Medical Device Device Registration Services
Freyr Expertise
- Regulatory Due-Diligence
- Device Documentation
- 513(g) support
- 510(k) Registration
- De-Novo Request for Classification
- PMA Registration
- 21 CFR 820 compliance
- BIMO Audit support
- MDSAP Compliance
- Labeling support
- Publishing and Submission support
- U.S. Agent
- Q-Submission meetings
- RFD and Pre-RFD Meetings
- Small Business Certification
- Establishment Registration and Device Listing
- Regulatory Compliance for Radiation medical Devices
- Post Approval Change Management
- Post Market Surveillance
- UDI Compliance
- Regulatory Consultancy for addressal of deficiencies