
What is QMSR?
The FDA QMSR is a streamlined approach to be in line with ISO 13485:2016 requirements, which is an update to the former QSR structure. This alignment is crucial because it simplifies global compliance for the manufacturers specially those who are operating globally. This harmonization will allow companies to meet Regulatory requirements both in the US and other markets in a much more unified manner.
QMSR mandates improvements in risk management, device design, post market surveillance. This Update may add more complexity, but it also provides the manufacturers to standardize their quality procedures, which in turn increases the device safety, also increase better documentation, which can be vital during USFDA inspections.
Why the QSR to QMSR transition is crucial?
![]()
![]()
Alignment with global standards
This will help the manufacturers in harmonizing with the global QMS for medical device requirements by upgrading to QMSR, which reduces Regulatory redundancies for the medical device manufacturers operating globally.
![]()
Improved Quality, Safety and Effectiveness
This integration ensures that the devices are manufactured as per the global QMS requirements. This helps the manufacturer to improve the quality and safety of the devices.
![]()
Regulatory Compliance
This helps the manufacturers to adapt to the new requirements and this reduces the risk of non-compliance when the regulation is enforced on Feb 2, 2026.



Key changes for QSR to QMSR Transition:
![]()
Harmonization with the terminology of ISO 13485:2016
This ensures that manufacturers to adopt globally accepted standards.
![]()
Risk management:
It emphasizes on the risk management throughout the lifecycle of the medical device.
![]()
Device design and controls
Under FDA QMSR, design controls expanded to ensure that manufacturers fully account for user needs, safety of the device and performance criteria, which is a major focus area during USFDA inspection
![]()
Post Market Surveillance
Companies need to boost the post market monitoring system. This will expect manufacturers to gather information on the device safety and effectiveness which help in finding the issues quickly and resolving issue quickly.
![]()
Documentation and record keeping
The final rule stresses on the documentation which should be thorough recordkeeping is critical during the inspection.
Important dates
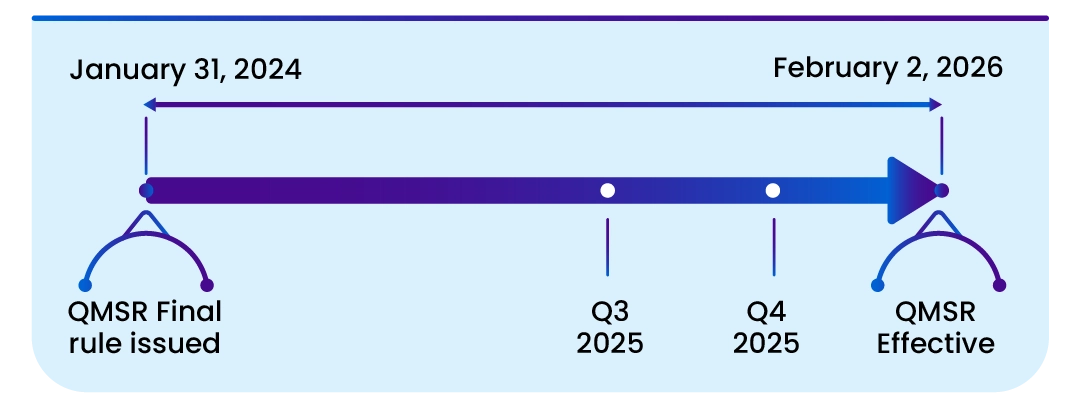
This rule is effective from February 2, 2026. The incorporation by reference of certain material listed in this rule is approved by the Director of the Federal Register on February 2, 2024.
Why should you adhere to FDA QMSR framework?
![Regulatory Intelligence (RI)]() QMSR will be enforced from Feb 2. 2026, which is mandatory for manufacturers to upgrade their QMS.
QMSR will be enforced from Feb 2. 2026, which is mandatory for manufacturers to upgrade their QMS.![A single partner for everything]() On time implementation and effective maintenance may reduce the likelihood of product recall and complaints.
On time implementation and effective maintenance may reduce the likelihood of product recall and complaints.![submission accuracy- ensuring]() Compliance to QMSR will may reduce the observations by US FDA which will not affect the product status in the Regulatory market.
Compliance to QMSR will may reduce the observations by US FDA which will not affect the product status in the Regulatory market.
 QMSR will be enforced from Feb 2. 2026, which is mandatory for manufacturers to upgrade their QMS.
QMSR will be enforced from Feb 2. 2026, which is mandatory for manufacturers to upgrade their QMS. On time implementation and effective maintenance may reduce the likelihood of product recall and complaints.
On time implementation and effective maintenance may reduce the likelihood of product recall and complaints. Compliance to QMSR will may reduce the observations by US FDA which will not affect the product status in the Regulatory market.
Compliance to QMSR will may reduce the observations by US FDA which will not affect the product status in the Regulatory market.Choose Freyr’s FDA QMSR consulting services, where our best-in-class consultants will guide you meticulously at every step of your device’s lifecycle to ensure seamless implementation of the QMSR requirements!
QMSR
- Classification of medical devices as per US-FDA.
- Creating documents as per 21 CFR 820.
- Gap analysis of existing Quality Management System (QMS) documents as per 21 CFR 820.
- Remediation plan for 21 CFR 820 compliance.
- Mock audits.
- A dedicated Quality Assurance (QA) team of experts on medical devices.
- Proven expertise in handling 21 CFR 820 compliance.
- Flexible project delivery models.