3 min read
The European Union Medical Devices Regulation (EU MDR) 2017/745 defines the scope of the sterile medical devices as devices that must be microorganism-free (bacteria, viruses, etc.). Class I devices with sterile function can also obtain a self-certification, but it differs in terms of requirements compared to other sub-categories of Class I devices.
The requirements within Class I devices vary greatly from manufacturing to placing the device on the market. The same applies to class I medical devices with sterile conditions. For instance, if it is a Class I device with a sterile function, the requirements for manufacturing and setting up a Quality Management System (QMS) would differ greatly compared to the class I devices with measuring functions.
This article aims to provide a concise checklist to consider while placing a Class I device with sterile conditions in the European Union. The checklist will support the manufacturers in making informed decisions concerning different aspects.
Dos!
Know the Scope Before Manufacturing
According to the EU MDR, medical devices with sterile functions must be developed and manufactured with appropriate procedures to ensure sterility until they are placed on the market. The devices can be reused only after appropriate cleaning, disinfection, and sterilization procedures.
Choosing the Correct Sterilizing Practices
There are different sterilization methods for a manufacturer to choose for sterilizing the device. The Ethylene Oxide (EtO) method is widely used to sterilize medical devices. The choice of sterilization must be appropriately justified. Some standards that manufacturers can follow for establishing, securing, and maintaining sterilization are EN ISO 14937, ISO TS 21387, ISO TS 13004, ISO 11137, and others.
Also, note that the documentation of the detailed sterilization method, in-process method, and data validation must be done accurately. Sterilization information, such as heating temperature, room conditions, process validations, etc., must be provided per the EU MDR requirements.
Know the Requirements and the Guidance to be Followed
One of the critical steps is documenting the technical requirements and knowing the standards accepted under EU MDR. For instance, EN 556 is the European standard for sterilized devices. EN 556 details the requirements for designating a medical device as sterile. Further, ISO TS 19930 specifies strategies for assuring the sterility of the devices. However, it is not yet adopted as an EU standard.
Know Your Route
In obtaining the CE mark, be informed about the involvement of Notified Bodies and the conformity assessment route that should be taken. Additionally, medical devices with sterile conditions are required to follow Annexes IX and XI.
The involvement of a Notified Body is required yet limited. In the case of sterile devices, the involvement of Notified Bodies will be with respect to maintaining and securing sterility conditions. For instance, the conformity assessments will involve microbiology assessments, aseptic methods, etc.
Don'ts!
Don’t Miss Out on the Packaging and Labeling Requirements

There are additional requirements for packaging and labeling compliance in sterile devices. While complying with the packaging and labeling requirements, one should not forget the information that is required to be affixed. The first and foremost step is to affix a sterile symbol (mentioned below) on the device labeling.
Information such as the declaration of sterile condition, method of sterilization, instruction in case the sterile packaging is damaged or opened unintentionally, etc. is to be included. Also, the shelf life of the sterile product and the re-sterilization process (if applicable) must be included. The identification number of a Notified Body should also be affixed to the device.
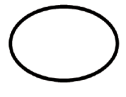
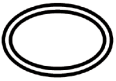
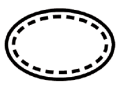
Some of the symbols that are required to be added (depending upon the applicability) in the labeling of sterile medical devices are:
Symbol | Indication |
| Sterile medical devices with sterilization method used as aseptic technique |
| Sterile medical devices with sterilization method used as ethylene oxide |
| Sterile medical devices with sterilization method used as dry heat or steam |
| Sterile medical devices with sterilization method used as vaporized hydrogen peroxide |
| Sterile medical devices with sterilization method used as radiation |
| The device cannot be sterilized again |
| The device is not sterilized |
| Do not use it if the package is damaged |
| Single sterile barrier system |
| Double sterile barrier system |
| Single sterile barrier system with protective packaging inside |
| Single sterile barrier system with protective packaging outside |


Don't forget the requirements for post-market placement
The sterile devices are required to maintain a record of adverse events in case of any occurrence. In this case, the recorded incidents, such as documents, are then required to be produced by the competent authorities upon request.
The EU MDR has specific requirements when it comes to self-certification. It is a complex procedure, the primary factor being different classes within the Class I device and their different aspects of requirements in obtaining a CE mark. Manufacturers with Class I devices sterile conditions should be aware of designating scope and specific requirements.
Do you wish to understand the various aspects of class I sterile devices in detail? Would you like to discuss this with a proven expert? Reach out to us.









