
The North American medical device market is one of the most attractive and highly regulated in the world. In the United States, medical device registration is overseen by the FDA, primarily through the Center for Devices and Radiological Health (CDRH), while in Canada, it is governed by Health Canada under a risk-based framework.
For manufacturers, navigating these markets can be complex. Both countries rely on risk-based medical device classification (Class I, II, III, and in Canada, Class IV), backed by robust evidence of safety and performance and a compliant quality management system (QMS). Programmes such as MDSAP and standards like ISO 13485, along with differing terminology, pathways, and documentation expectations, add further layers of complexity when planning entry into the US and Canadian markets.
Freyr helps manufacturers turn this complexity into a clear, actionable plan for the US and Canada market access. We understand how FDA requirements and Health Canada medical device regulations intersect, and we support you in aligning classifications, evidence, QMS, and dossiers so you can follow a single, efficient strategy for approvals, launches, and long-term compliance in both countries.
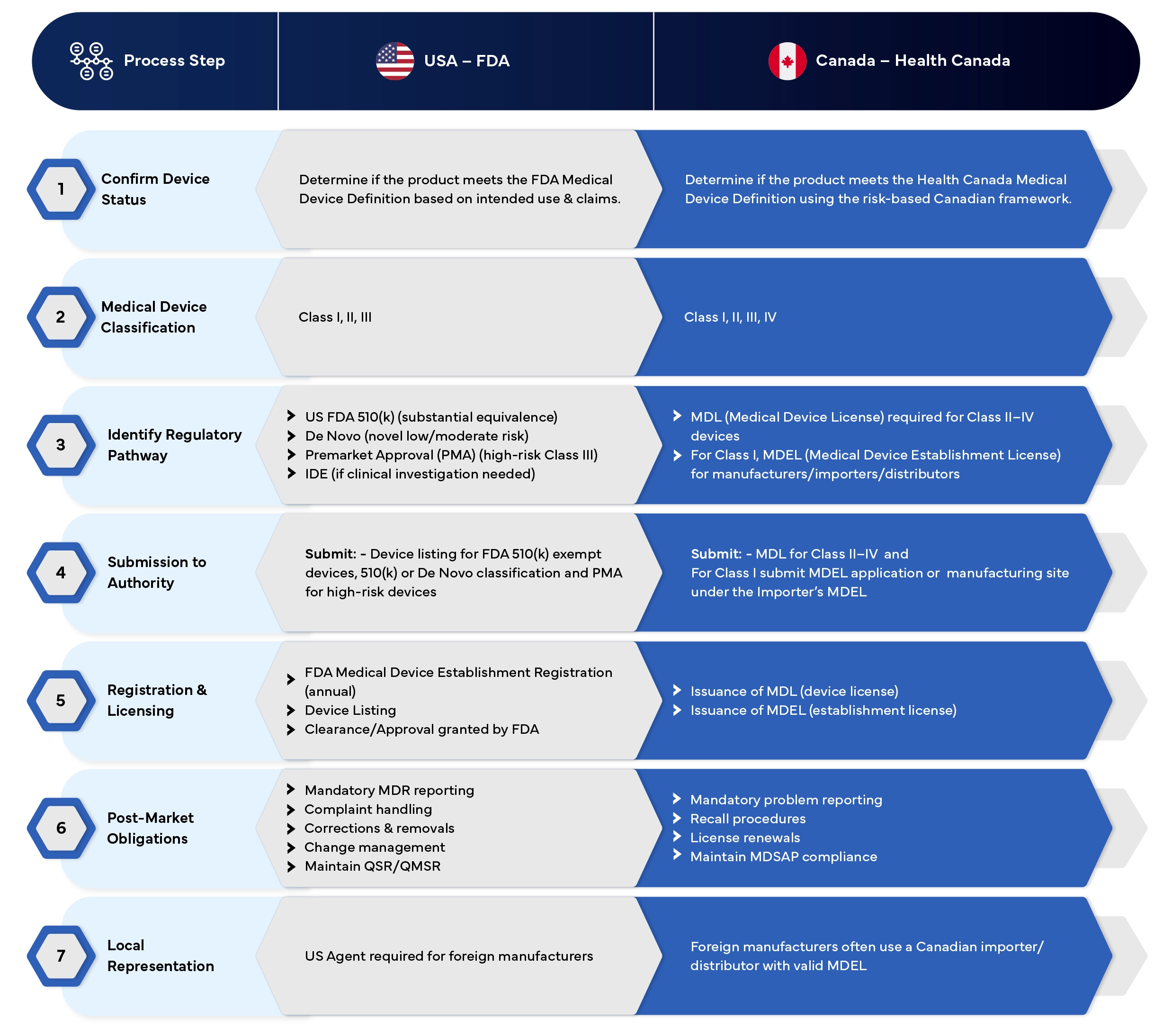
USA vs Canada
Medical Device Registration Process - Comparison Chart
Freyr’s Medical Device Registration Services
Medical Device Registration in The Americas
- Regulatory and Market Intelligence Support
- Product Classification as per Regulatory requirements
- Medical Device Registration USA -513 (G) & (Right Pathway among 510 (K)/De-NOVO/PMA/IDE)
- Pre-Submission Meeting with Health Agencies
- Regulatory Support for Product Documents such as, Design History File (DHF)
- Gap Analysis of the Technical Documents & Good Manufacturing Practice (GMP) Systems
- Regulatory Support for Technical File Compilation as required by respective Registration Pathway
- USFDA Quality System Compliance Strategy (21 CFR 820)
- Medical Device Single Audit Program (MDSAP) compliance services
- Medical Device Registration Canada-Medical Device License (MDL) and Medical Device Establishment License (MDEL) Registration
- Health Agency Liaising and support
- In-Country Representation – U.S. Agent and Sanitary Responsible Person Services
- Post Approval Compliance activities

- Successful submissions for varied class of devices ranging from software to sutures
- Local affiliate access to meet the challenges of authority and language-specific requirements
- Dedicated and expert personnel to provide Medical Device and IVD Regulatory support
- Cost-effective model for In-country or Legal representation support
- Have successfully delivered various projects across U.S. and Canada
- Headquartered in the U.S.

Why Partner with Freyr?
- End-to-end North America regulatory expertise, supporting everything from FDA and Health Canada pre-market notification (submissions) to post-market surveillance and lifecycle compliance.
- Proven success across 1500+ global medical device registrations, spanning Class I–III devices in the US and Class I–IV devices in Canada.
- On-ground presence through US Agent services and Canadian regulatory support, backed by globally distributed expert teams for seamless execution.
- Integrated US & Canada strategy that streamlines evidence, documentation, and quality requirements to reduce cost, duplication, and approval timelines.
- Direct engagement with FDA and Health Canada, ensuring clear communication, timely responses, and proactive alignment with regulatory expectations.
- Trusted by 470+ medical device manufacturers worldwide, from start-ups to global enterprises, as their long-term North America regulatory partner.
Celebrating Customers Success
Medical Devices
Registration and LR Support
Global
Freyr has been an indispensable partner in achieving rapid global scalability for our Software as a Medical Device (SaMD) business. As a startup, acquiring expertise in worldwide regulations is cost-prohibitive. Freyr's competitive pricing and tailored services allowed us to get that expertise at a fraction of the cost of full-time resources. Their team's responsiveness and adaptability to project priorities have greatly facilitated our progress. We recommend Freyr to any company seeking expert guidance and support in the medical device Regulatory domain.
Arie Henkin
VP - Quality and Regulatory, Australia -based, Leading SaMD Company
Medical Devices
Swiss Rep Services
Japan and Switzerland
I genuinely enjoy my time working with Freyr, and I view them as a truly valuable asset and extension to my own team. They are dependable and accurate, and their pricing is competitive. Beyond that, I won’t hesitate to collaborate with Freyr again.
Darren Mansell
Regulatory Affairs Manager, UK-based, Global Medical Device Design and Manufacturing Company
Medical Devices
Registration and AR Services
Malaysia and Indonesia
Freyr provides a reliable service with expertise across many countries. I can rely upon Freyr to provide the information necessary to make an informed decision before entering a formal scope of work agreement. Once a project is underway, the Freyr team acts professionally to execute the work with excellent communication of progress.







