

3 min read
Undertaking a clinical trial is a major step in the development of a drug product or medical device. A Clinical Trial Application (CTA) is required for pharma companies or sponsors to seek Regulatory authorities' approval to begin human studies. As clinical research becomes more global and complex, understanding the evolving international practices, critical submission steps, and the advantages offered by a Regulatory Affairs partner/s is crucial for sponsors and research organizations alike.
Clinical Trial Application (CTA) and its Sections:
A Clinical Trial Application (CTA) is the Regulatory dossier that sponsors must submit to national Regulatory authorities (NRAs) before starting a clinical study involving human participants. CTAs are mandatory for most interventional drug, device, or biologic trials. They ensure the trial is scientifically sound, ethically appropriate, and aligned with local and international guidelines—such as the International Council for Harmonization (ICH) Good Clinical Practice (GCP) standards and national requirements.
Key sections in a typical CTA include (not limited to):
- Cover letter and application forms
- Clinical study protocol and investigator’s brochure
- Product dossier and Good Manufacturing Practice (GMP) evidence
- Ethics and Institutional Review Board (IRB) approvals
- Insurance and subject protection documentation
- Data safety monitoring plan and financial declarations
Global Practices in CTA Submissions
CTA processes and requirements differ across regions, but several common principles are emerging:
1. Harmonization of Formats and Standards
- Many countries use the ICH Common Technical Document (CTD), streamlining multinational study submissions. Its modular structure supports parallel review and simplifies updates.
- Local adaptations are common, such as in the Asia-Pacific and Western Pacific regions, where countries often blend the CTD with national forms, review feedback, language needs, or bridging studies.
2. Regulatory Consultation
- Early engagement with agencies—such as the US FDA, EU national authorities, and agencies in Japan, China, and key emerging markets—is encouraged, especially for global and multi-regional clinical trials (MRCTs). Scientific advice meetings reduce future rejections and steer protocol optimization.
3. Collaborative and Accelerated Pathways
- Agencies increasingly accept “reliance” or collaborative assessments, where they leverage reviews or approvals by stringent Regulatory authorities, boosting speed and consistency.
- Fast-track or expedited review options may be available, especially during health emergencies or for breakthrough therapies.
4. Ethics Review Integration
- In the EU and many other regions, ethics and Regulatory reviews may occur in parallel or through coordinated platforms to avoid separate submissions and shorten start-up times.
5. Transparency and Publication
- It’s now standard to register trials in recognized databases before approval, and many countries publicly report CTA approval statuses, contributing to global transparency and best practices.
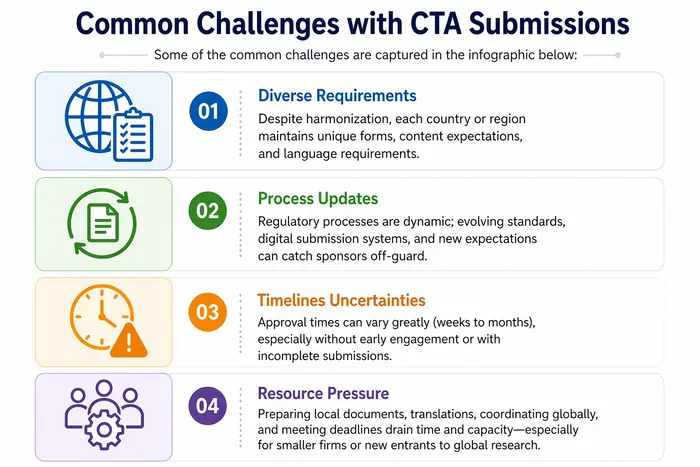
How Can a Partner Like Freyr Help?
A trusted Regulatory Affairs (RA) partner like Freyr provides seamless support and risk mitigation throughout the CTA process. Here’s why engaging such an expert sets sponsors up for success:
1. Up-to-date Global Regulatory Intelligence
- We continuously monitor Regulatory changes, regional interpretations, and submission practices worldwide. This ensures every CTA is built around the most current requirements—minimizing queries, rejections, and costly trial delays.
2. Dossier Preparation and Review
- Freyr’s specialists develop and compile CTAs using the latest ICH CTD formats and tailored national templates. They coordinate document translations, organize dossier responses, and ensure uniform quality and GCP compliance for each submission.
3. Regulatory Consultation and Agency Liaison
- Freyr can represent sponsors in pre-CTA scientific advice meetings, handle agency questions post-submission, and support communications with both authorities and Ethics Committees.
4. Global Project Management
- With deep multicultural teams and robust project management tools, Freyr coordinates simultaneous submissions, tracks country-by-country progress, and drives alignment for MRCTs (Multi-Regional Clinical Trials).
5. Reliance and Accelerated Review Strategy
- Freyr helps sponsors leverage reliance frameworks, prepare abridged dossiers for waived or fast-tracked reviews, and align submission data for maximum Regulatory acceptance.
6. Audit and Change Control Readiness
- The partner ensures that sponsors’ documentation and procedures meet audit standards, facilitating swift regulatory approval and smooth inspections.
Conclusion
CTA submission is a complex but essential gateway for clinical research, with global and region-specific requirements. Global trends include CTD harmonization, parallel ethics and Regulatory review, reliance mechanisms, and new transparency norms. Sponsors face notable challenges in meeting diverse requirements, keeping pace with Regulatory changes, and managing global timelines. In such a scenario, a Regulatory Affairs partner such as Freyr provides end-to-end expertise in Regulatory intelligence, dossier preparation, agency interactions, accelerated pathways, and audit-readiness—substantially increasing the odds of timely, successful clinical trial initiations.









