

2 min read
With the vision of global device traceability, South Korea’s Ministry of Food and Drug Safety (MFDS) has suggested/mandated? a new Regulatory requirement, for incorporating Unique Device Identification system (UDI) on medical devices. Medical device registration and UDI are integrated procedures, and UDI is a prerequisite for establishing medical devices in the Korean market. To ensure patient safety, the MFDS introduced the Integrated Medical Device Information System (IMDIS) in 2016, a platform to regulate the traceability of medical devices in South Korea.
According to Article 2 of the Medical Device Act specifying UDI medical device regulations, a UDI system includes the numbers and barcodes indicated on the container and the package, etc., of medical devices in a standardized system to identify and manage them thoroughly and effectively. MFDS expects manufacturers to register the information within the IMDIS platform. IMDIS is used to electronically link the information system related to medical devices.

Source: Easy Medical Device
UDI consists of a machine-readable barcode that includes:
- Device Identifier- a combination of figures or alphabets uniquely generated for each product in the UDI
- Product Identifier- a combination of figures or letters generated by the production unit in the UDI. It includes the manufacturing number (Lot, serial number), date of production, and the information on the product version
Timelines to Incorporate UDI Requirements in the Packaging of Medical Devices:
As a part of the Medical Device Act revision in 2016, IMDIS was introduced to understand and carry out a systematic tracing of devices. After successful enforcement of the IMDIS platform, incorporation of UDI in the packaging was conducted in a phase-wise manner, wherein the UDI was made mandatory as shown in the below pictorial.
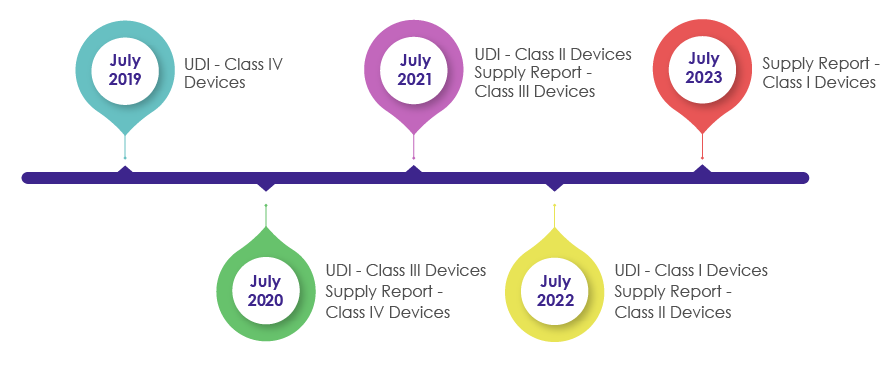
MFDS needs medical device manufacturers, vendors, and suppliers to record the supplied devices data and provide the supply information (which includes Supplier, Lot Number, Packaging Unit, Quantity, Date, Unit price by sale price) in the supply reporting in the UDI.
Process of Generating a UDI:
Medical device manufacturers or importers shall assign a UDI to their devices by model name and package unit under Article 3(2) before shipping medical devices that are permitted, certified, or reported. When the UDI is marked as a barcode, the GS1 International Standard System shall be used. For the UDI-DI, a Global Trade Item Number (GTIN) code shall be used, while for the UDI-PI, GS1 Application Identifiers (AI) shall be used.
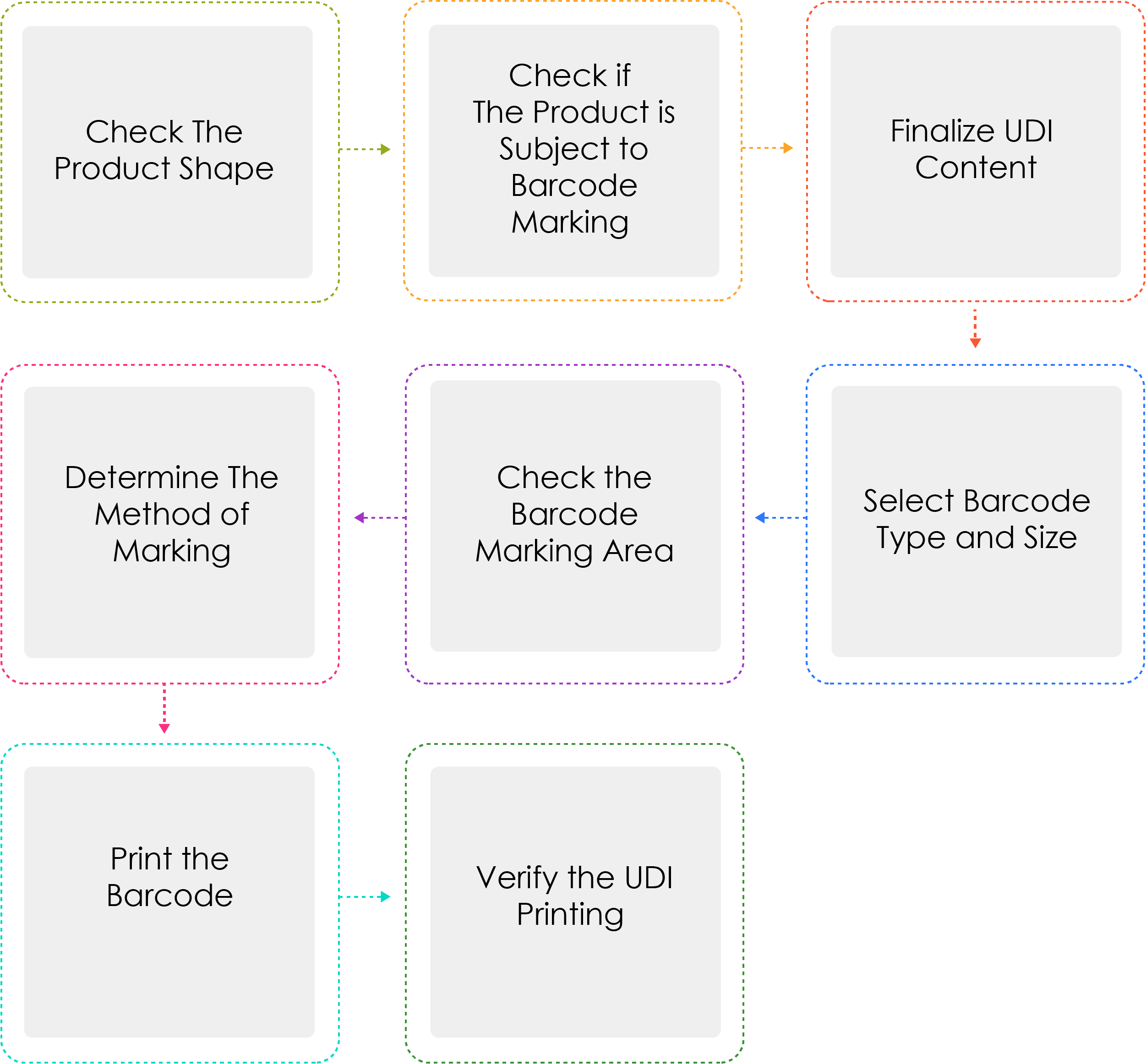
Why is Determine the method of marking written thrice ?
With the implementation of UDI compliance requirements, MFDS can streamline the pre-market activities (i.e., QMS conformity, Clinical trial assessment, and overall approval) as well as the post-market activities (i.e., AE reporting, recalls) of the devices. MFDS’ conscious attempt to trace and monitor the devices will ensure the safety and effectiveness of the devices in the South Korean market.
To know more about UDI compliance and the medical device registration process in South Korea, reach out to Freyr.









