
3 min read
Labeling is an integral part of marketing medical devices. The label is a piece of information affixed with the device and/or packaging in a human-readable format. The main purpose of labeling is to provide safety information to users who can be healthcare professionals, consumers, or any other relevant person.
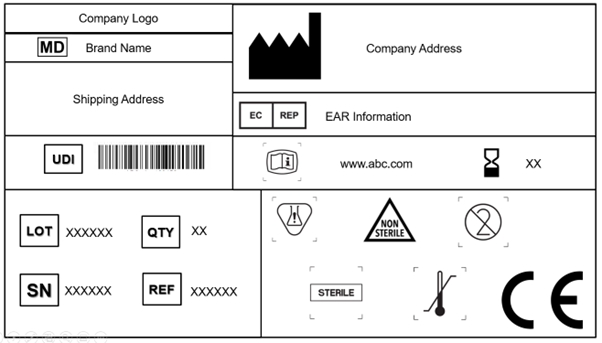
All the global Regulatory authorities have certain labeling requirements. Likewise, the EU has detailed the labeling requirements in Chapter III under Annex I of the EU Medical Devices Regulations (EU MDR) 2017/745. The foremost important thing to note is to include all the symbols covering the required information in the labeling of the device and the documents (booklets, manuals, IFUs, etc.) accompanied by it.
Some of the critical labeling considerations to be noted while complying under EU MDR 2017/745 are-
1. Medical Device Labeling Symbology
Every manufacturer is required to incorporate the medical device symbol, which states that the product supplied to the EU market is a medical device. It is mandatory to affix this symbol on the device and all levels of packaging. In addition, the label should project the trade name and the device’s original name.
2. Special Devices
In case the product is a special or custom device, the status of the same should be mentioned on the labeling. For instance, if the product’s intent is only for clinical investigation, the label should explicitly say so.
For the devices with absorbent materials or which may locally disperse in the human body, the labeling should mention the composition of the material and quantitative details on the key constituents.
Even explicit labeling is required in the case of single-use devices and sterile devices. For the reprocessed devices, the labeling should mention the number of times they can be reprocessed, the number of times they have been reprocessed so far, and the sterilization method used.
3. Presence of Toxic Substances
The declaration of the presence of CMR (carcinogenic, mutagenic, toxic for reproduction) substances and endocrine disrupting substances is mandatory on the labels if the concentration is more than 0.1% w/w. The list of such substances must be affixed to the device and/or packaging.
Further, a label on the presence of blood and tissue derivatives (even when contained in the medicinal substance of the combination device) must be affixed to the devices.
4. Harmonized Standards
The EU MDR 2017/745 recognizes and accepts the ISO 15223-1: 2021. The document determines the symbols to be used in the Labeling of medical devices and their packaging. Chapter 3 (23.1,h) of Annex I in the EU MDR specifies that internationally recognized symbols can be used, and in the case of regions where these symbols are not recognized, the description of the same is required to be supplied in a document along with the device.
5. UDI
Article 27, 28, 29, and Annex VI (A, B, C) detail out rules and regulations for the UDI. The label is now required to contain a UDI carrier [Automated Identification for Data Capture (AIDC) and Human-Readable Interpretation (HRI) representation of the UDI] on the device and higher packaging levels as well. The device’s higher packaging (excluding the shipping packages) will have its own UDI carrier.
6. Electronic Information for Use (eIFU)
Web address (URL) in the form of eIFUs can also be placed on the medical device labeling along with the paper IFUs. The eIFUs can be used in the case of implantable, active implantable, fixed medical devices and software (intended for laymen as well).
7. Information of Economic Operators (EOs)
The label usually contains the information of the manufacturer. However, in the case of foreign manufacturers, the authorized representative’s information should be placed on the commercial labels.
8. Warnings and Precautions
The warnings and precautions must be mentioned on the device’s label. The information on this aspect can be kept minimum, and details of the same can be provided on the IFU.
The manufacturers are also required to accustom to the country-specific Labeling requirements. The language requirement is dependent on the EU Member State. It can heavily impact the device’s Labeling, IFUs, and packaging in terms of time and costs.
These additional requirements can further add up to the manufacturer's burden with the existing labeling process complexity. The failure on the same can become very costly, involving product recalls and subsequent steps for Corrective and Preventive Action (CAPA).
Are you looking for assistance on labeling as per the EU MDR? Freyr offers comprehensive services in labeling medical devices. Feel free to connect with our Regulatory experts now at – sales@freyrsolutions.com









