
Aperçu des services de conformité EU MDR
Le Règlement de l'UE relatif aux dispositifs médicaux (EU MDR) est entré en vigueur le 26 mai 2021, après une période de transition de trois ans et une prolongation d'un an supplémentaire due à la pandémie de COVID-19. Les dispositifs mis sur le marché de l'UE doivent désormais se conformer à ces réglementations et être certifiés CE selon l'EU MDR par les organismes notifiés accrédités en vertu de ces réglementations. Cependant, les dispositifs déjà certifiés CE selon la directive de l'UE sur les dispositifs médicaux (EU MDD) bénéficient de périodes de grâce avant de devoir se conformer entièrement aux exigences de l'EU MDR. Pendant cette période de grâce, les dispositifs certifiés selon l'EU MDD et l'EU MDR coexisteront sur le marché avec un statut égal et sans discrimination. Freyr propose des services de conformité EU MDR inégalés pour aider les entreprises de dispositifs médicaux à respecter les exigences de l'EU MDR dans les délais impartis.
Calendrier de transition et nouvelles classifications des dispositifs
Le règlement européen sur les dispositifs médicaux (MDR) sera pleinement en vigueur dans tous les Member States de l'UE et les États de l'Association européenne de libre-échange (AELE) à partir de mai 2021 et accorde aux fabricants une période de transition de 4 ans pour une certification MDR UE complète.
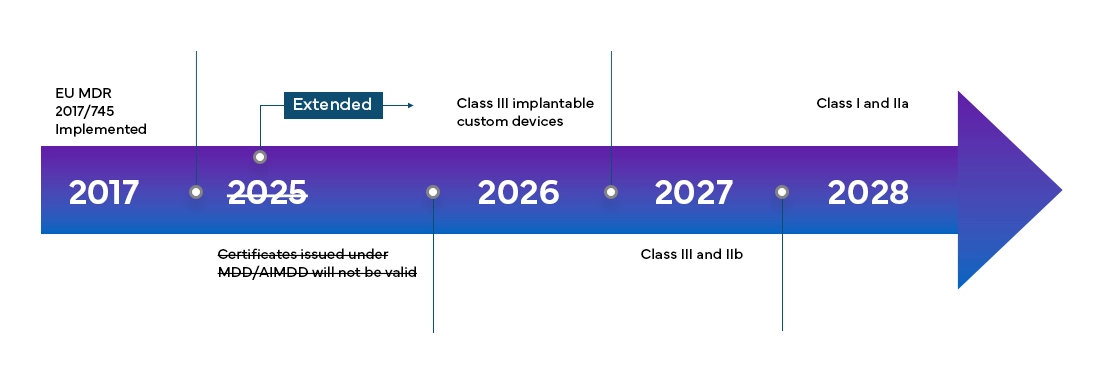
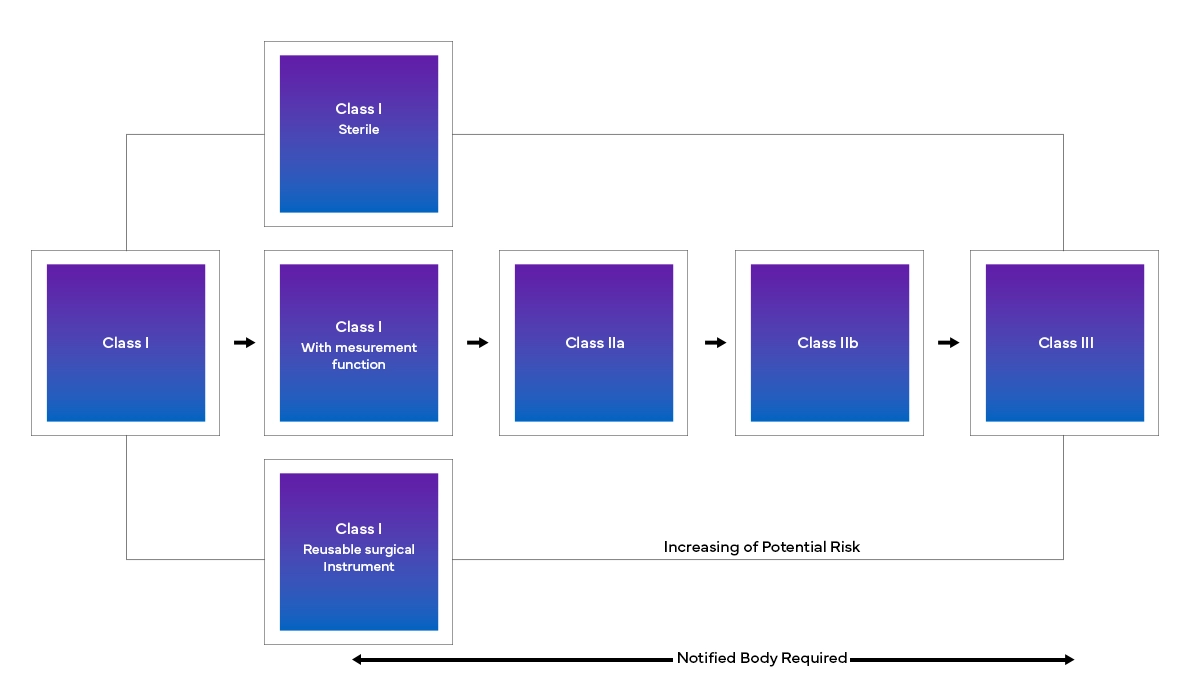
La nouvelle Réglementation européenne sur les dispositifs médicaux (MDR), comme constaté, a également apporté des changements au système de classification des dispositifs existant, tels que :
De l'identification des modifications exactes à apporter à leur mise en œuvre en temps réel, les fabricants peuvent être confrontés à une série de défis pour se conformer aux exigences du règlement européen sur les dispositifs médicaux (EU MDR). De la compréhension de la nouvelle structure à la classification précise d'un dispositif, en passant par la collecte et la soumission de toutes les données, une approche réglementaire plus détaillée et transversale sera nécessaire pour les fabricants afin de faire face aux nouvelles réglementations européennes sur les dispositifs médicaux. Grâce à une analyse d'écart rigoureuse, Freyr aide ses clients à évaluer la situation actuelle et leur fournit les mesures réglementaires nécessaires pour la transition et la conformité au règlement européen sur les dispositifs médicaux (EU MDR).
Obtenez des conseils d'experts sur votre conformité au EU MDR
Services de conformité EU MDR
- Élaboration d'une stratégie claire de mise en œuvre du Règlement sur les dispositifs médicaux (MDR)
- Comprendre la nouvelle législation, réaliser une analyse des écarts par rapport aux systèmes de gestion de la qualité (SGQ) et aux processus actuels en place
- Élaboration d'un plan détaillé avec une approche transversale pour déterminer les aspects du système qualité qui devront être modifiés en conformité avec le nouveau Règlement européen sur les dispositifs médicaux
- Formation de plusieurs équipes pour l'analyse de la portée du produit, la classification, la gestion du SMQ, etc., au sein de l'organisation, avec un point de contact unique dans chaque équipe
- Allocation et planification des ressources
- En tenant compte de l'interaction de votre SMQ avec d'autres réglementations et en saisissant cette opportunité pour rationaliser les processus, tout en permettant une flexibilité pour intégrer les changements futurs.
- Analyser les données de test existantes et vérifier toute exigence supplémentaire imposée par le MDR
- Coordination des attentes et du plan de transition avec vos organismes notifiés de l'UE
- Analyse des écarts pour les dispositifs médicaux existants, de la directive européenne sur les dispositifs médicaux (EU MDD) au règlement européen sur les dispositifs médicaux (EU MDR).
- Soutien End-to-End pour l'élaboration du rapport d'évaluation clinique (REC), y compris la recherche bibliographique conformément aux lignes directrices du règlement européen sur les dispositifs médicaux (EU MDR)
- Services End-to-End pour les rapports de surveillance après commercialisation (RSAC), les rapports périodiques actualisés de sécurité (RPAS) et le résumé des caractéristiques de sécurité et des performances cliniques (SSCP)
- Renforcement des équipes réglementaires avec des options de déploiement sur site et à distance
- Services de Représentant autorisé européen (EAR)
- Conformité au MDR et assistance pour la soumission aux Organismes Notifiés
- Veille réglementaire concernant le processus d'importation des différents marchés réglementés
- Conformité SMQ et audits blancs
- Système de gestion documentaire et outil pour les entreprises MDR
- Classification et reclassification des dispositifs en fonction du risque
- Mise en œuvre et conseil UDI
- Services de surveillance après commercialisation conformes au Règlement de l'UE sur les dispositifs médicaux
- Conseil en gestion des risques ISO 14971
- Formations internes et en ligne
- Personne responsable des services et de l'assistance en matière de conformité réglementaire
- Identification des organismes notifiés au titre du RDM

Pour un support réglementaire End-to-End concernant l' EU MDR, contactez Freyr.







