
Garantir une conformité sans faille aux réglementations et aux procédures d'enregistrement britanniques relatives aux dispositifs médicaux
Les fabricants étrangers doivent se conformer au règlement britannique de 2002 sur les dispositifs médicaux (tel que modifié). Tous les dispositifs médicaux et les dispositifs de diagnostic in vitro doivent être enregistrés auprès de la MHRA leur mise en vente au Royaume-Uni. Les fabricants non britanniques sont tenus de désigner une personne responsable au Royaume-Uni (UKRP) pour les représenter. La MHRA des exigences strictes en matière de documentation, d'étiquetage et de notification des incidents afin de garantir la sécurité des patients et la traçabilité des produits.
Il peut s'avérer complexe de s'y retrouver parmi les nouvelles exigences proposées dans les conclusions de la consultation pour le Royaume-Uni, notamment en ce qui concerne le marquage UKCA, les exigences en matière de données et les attentes après la mise sur le marché. De nombreuses entreprises sont confrontées à des difficultés liées à des dossiers techniques incomplets, à des classifications divergentes des dispositifs médicaux et à une connaissance limitée du système d'enregistrement en ligne des dispositifs médicaux (DORS). Les mises à jour réglementaires et l'évolution MHRA compliquent encore davantage la planification et l'affectation des ressources.
Freyr simplifie chaque étape de votre entrée sur le marché britannique. Nous gérons les demandes DORS, agissons en tant que votre UKRP, veillons à ce que tous les documents soient prêts et comblons les lacunes en matière de conformité lors de la transition du marquage CE vers le marquage UKCA. Nos experts en réglementation vous accompagnent dans l'évolution MHRA , vous aidant ainsi à obtenir un accès au marché conforme, rapide et rentable.
Procédure étape par étape pour l'enregistrement des dispositifs médicaux au Royaume-Uni
L'enregistrement d'un dispositif médical au Royaume-Uni comporte plusieurs étapes bien définies. Voici comment Freyr gère l'ensemble du processus pour vous :
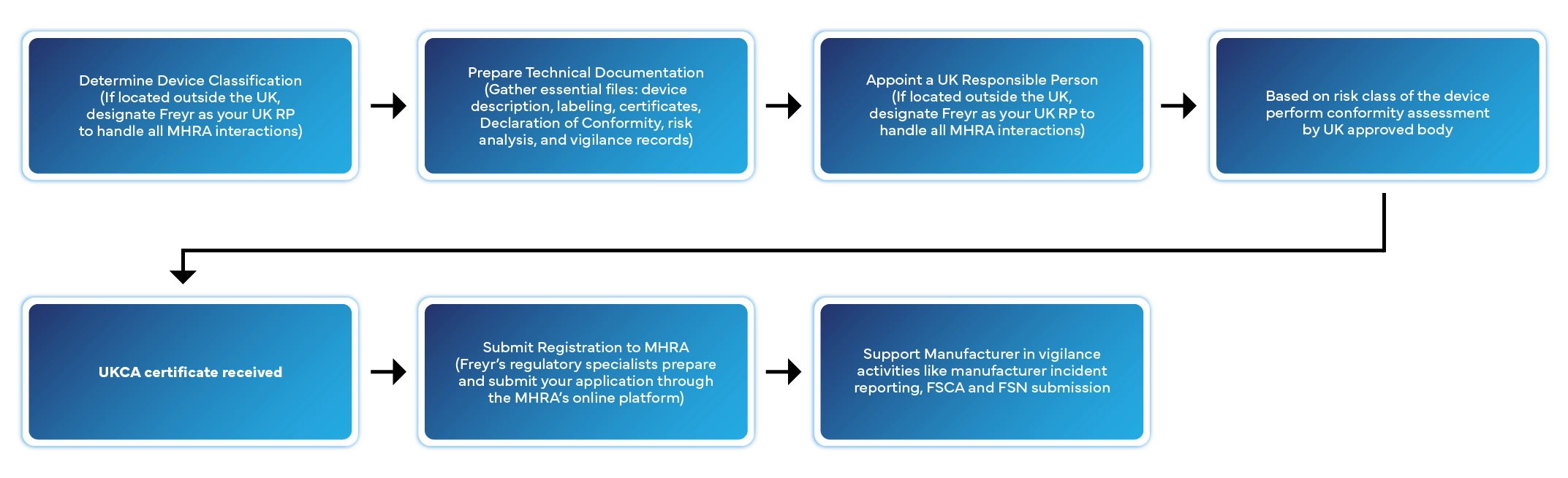
Délai de traitement habituel : 2 à 6 semaines, selon la catégorie du dispositif et l'exhaustivité du dossier.
Principales offres de Freyr Medical Device UK

Enregistrement des dispositifs: Freyr gère l'ensemble MHRA via la plateforme DORS, garantissant ainsi la conformité des demandes, l'exactitude des données et l'obtention rapide des autorisations pour toutes les catégories de dispositifs.

Responsable désigné au Royaume-Uni En tant que UKRP désigné, Freyr vous représente auprès de la MHRA, gère toutes les communications et veille au respect constant des réglementations ainsi qu'à la surveillance continue.

TF / Compilation de dossiers: Freyr compile des dossiers techniques et des documents conformes aux exigences réglementaires britanniques relatives au marquage UKCA, garantissant ainsi la préparation aux audits, aux inspections et aux soumissions.

Assistance en matière de systèmes de gestion de la qualité Nous vous aidons à mettre en place et à maintenir des systèmes de gestion de la qualité ISO 13485, adaptés aux MHRA du règlement britannique sur les dispositifs médicaux (MDR) et MHRA santé MHRA .

Assistance à la rédaction de documents réglementaires: Freyr assure la préparation par des experts de rapports d'évaluation clinique (CER), de plans de surveillance post-commercialisation (PMS), de rapports de sécurité périodiques (PSUR) et de documents relatifs à la gestion des risques, en garantissant la clarté technique et la conformité réglementaire.

Étiquetage et conformité Notre équipe veille à ce que vos étiquettes, vos notices d'utilisation et vos emballages respectent les exigences britanniques en matière d'étiquetage et de langue (UKCA), en garantissant la cohérence et la conformité.

Surveillance post-commercialisation Freyr apporte son soutien aux activités de surveillance post-commercialisation, notamment la notification des effets indésirables, les déclarations de vigilance et MHRA , afin de garantir le maintien de l'accès au marché.
Offre de services de Freyr UK en matière de responsable désigné (UKRP)
Freyr agit en tant que représentant autorisé au Royaume-Uni ( UKRP) et veille au respect total de la réglementation britannique de 2002 sur les dispositifs médicaux (UK Medical Devices Regulations 2002) MHRA. Nous représentons votre entreprise au Royaume-Uni et assurons toutes les communications avec la MHRA.
- Enregistrement des dispositifs auprès de MHRA
Freyr agit en tant que votre responsable désigné au Royaume-Uni (UKRP) pour gérer l'intégralité du processus MHRA via la plateforme DORS, garantissant ainsi que chaque dispositif est correctement répertorié, vérifié et autorisé à la vente au Royaume-Uni. - Documentation et assurance de la conformité
Nos experts en réglementation veillent à ce que votre déclaration de conformité, votre documentation technique et vos certifications de produits soient disponibles, et en conservent une copie afin de pouvoir les transmettre à MHRA sur simple demande. - Réponse aux MHRA
Freyr gère directement, MHRA votre nom, toutes les communications et clarifications avec la MHRA , garantissant ainsi des réponses rapides et précises aux demandes réglementaires ou aux examens post-commercialisation. - Vigilance et communication des incidents
En tant que représentant autorisé UKRP, Freyr assure la liaison principale pour toutes les questions liées à la sécurité. Nous assurons la coordination entre les fabricants, les professionnels de santé, les patients et la MHRA les événements indésirables, en veillant à ce que ceux-ci soient correctement signalés et qu'des mesures correctives soient prises. - Préparation aux inspections et aux audits
Freyr conserve l'ensemble de la documentation et de la correspondance requises pour MHRA et les audits MHRA . Notre équipe veille à ce que les dossiers techniques, l'étiquetage et les registres post-commercialisation soient facilement accessibles.
Prenez rendez-vous avec nos experts dès aujourd'hui
Pourquoi collaborer avec Freyr ?
- Une expertise End-to-end , allant de l'enregistrement préalable à la mise sur le marché à la vigilance post-commercialisation, couvrant toutes les étapes de la conformité.
- Une expérience éprouvée, avec plus de 1 500 enregistrements d'appareils menés à bien dans diverses catégories.
- Une présence locale au Royaume-Uni, avec des spécialistes de la réglementation sur place à Reading, soutenus par des équipes opérationnelles internationales.
- Une planification de transition sur mesure qui apporte un soutien stratégique pour passer du marquage CE au marquage UKCA en toute fluidité et à moindre coût.
- Une communication transparente grâce à un contact direct avec MHRA à des mises à jour proactives sur la conformité destinées aux clients.
- Reconnue par des marques internationales, Freyr accompagne plus de 470 fabricants à travers le monde en tant que partenaire en matière de réglementation.
Foire aux questions (FAQ)
01. En quoi consiste la procédure d'enregistrement des dispositifs médicaux au Royaume-Uni ?
La procédure d'enregistrement des dispositifs médicaux au Royaume-Uni consiste à notifier la MHRA Agence de réglementation des médicaments et des produits de santé) concernant votre dispositif avant sa mise sur le marché en Grande-Bretagne. Les fabricants doivent fournir les coordonnées de leur entreprise, la classification du dispositif et la documentation technique. Les fabricants non britanniques doivent également désigner un responsable au Royaume-Uni (UK RP) chargé de gérer l'enregistrement et les communications relatives à la conformité au quotidien.
02. Qui doit désigner un responsable au Royaume-Uni (UK RP) ?
Tout fabricant établi en dehors du Royaume-Uni doit désigner un responsable au Royaume-Uni avant de mettre des dispositifs sur le marché britannique. Ce responsable au Royaume-Uni fait office d'interlocuteur réglementaire du fabricant et veille à ce que l'ensemble de la documentation technique, des déclarations et MHRA soient correctement gérées. Les fabricants établis au Royaume-Uni peuvent communiquer directement avec la MHRA un responsable au Royaume-Uni.
03. Quels sont les documents requis pour MHRA ?
La MHRA certains documents essentiels, notamment la déclaration de conformité, la description et la classification du dispositif, les coordonnées du fabricant et les informations relatives à l'étiquetage. Pour les dispositifs à haut risque, les dossiers techniques et les données cliniques peuvent également faire l'objet d'un examen. Disposer d'une documentation complète et prête pour un audit permet d'accélérer les procédures d'autorisation et de faciliter les inspections post-commercialisation menées par la MHRA les mandataires.
04. Combien de temps dure la procédure MHRA ?
En règle générale, MHRA prend entre 2 et 6 semaines, en fonction de la classe du dispositif, de l'exhaustivité du dossier et de la présence ou non d'un responsable au Royaume-Uni. Les dispositifs simples de classe I peuvent être traités plus rapidement, tandis que les produits complexes ou à haut risque peuvent nécessiter plus de temps en raison de la validation supplémentaire des données et de l'examen approfondi de la documentation technique.
05. Quelle est la différence entre le marquage CE et le marquage UKCA ?
Le marquage CE atteste de la conformité aux réglementations de l'Union européenne, tandis que le marquage UKCA (UK Conformity Assessed) s'applique aux dispositifs commercialisés en Grande-Bretagne. Depuis le Brexit, le marquage UKCA a remplacé le marquage CE sur le marché britannique, bien que ce dernier continue d'être accepté à titre temporaire. L'Irlande du Nord continue de reconnaître les marquages CE et CE UKNI en vertu des règles d'alignement sur l'UE.
06. Quelles sont les responsabilités après la mise sur le marché, une fois l'enregistrement effectué ?
Une fois l'enregistrement effectué, les fabricants et les responsables au Royaume-Uni doivent assurer la surveillance post-commercialisation, signaler les incidents indésirables, informer MHRA toute modification apportée au produit ou à son étiquetage, et renouveler les enregistrements si nécessaire. Ils doivent également conserver la documentation technique pendant au moins dix ans. Ces mesures garantissent le respect continu de la réglementation et préservent la santé des patients tout au long du cycle de vie du dispositif.
07. À quelle fréquence faut-il mettre à jour les informations MHRA ?
Les informations d'enregistrement doivent être mises à jour sans délai en cas de modification de la classification du dispositif, de son étiquetage, de son site de fabrication ou de l'entité responsable. La MHRA de la personne responsable au Royaume-Uni ou du fabricant qu'ils procèdent à des mises à jour immédiates afin de garantir l'exactitude des registres de mise sur le marché. Des audits internes réguliers permettent de s'assurer que vos données restent à jour et conformes à l'évolution de la réglementation.
08. Quelles sont les sanctions prévues en cas de non-respect de MHRA ?
Le non-respect des exigences britanniques en matière d'enregistrement des dispositifs médicaux ou de surveillance post-commercialisation peut entraîner des avertissements réglementaires, le retrait du produit, des amendes ou des poursuites judiciaires. La MHRA est MHRA habilitée à suspendre ou à révoquer l'autorisation de mise sur le marché. Une approche proactive en matière de conformité et la désignation d'un responsable qualifié au Royaume-Uni aident les fabricants à éviter les sanctions et à garantir la continuité de la commercialisation.
09. Comment Freyr simplifie-t-il l'enregistrement des dispositifs médicaux au Royaume-Uni ?
Freyr vous offre end-to-end , allant de la désignation de votre responsable au Royaume-Uni (UKRP) à la gestion MHRA , de la documentation technique et de la conformité continue. Nos consultants expérimentés simplifient le processus, raccourcissent les délais d'autorisation et contribuent à garantir un accès continu au marché.







