
Aperçu du protocole de recherche documentaire et de la revue de la littérature sur les dispositifs médicaux et les produits de diagnostic in vitro (IVD)
Dans le domaine complexe des dispositifs médicaux et des dispositifs médicaux de diagnostic in vitro (IVD), un protocole de recherche documentaire bien structuré pour les dispositifs médicaux est bien plus qu'un simple exercice de recherche : il s'agit d'une exigence essentielle pour se conformer au EU MDR et au règlement européen IVDR 2017/746.
Une analyse approfondie de la littérature médicale vient étayer les évaluations cliniques et de performance, les activités post-commercialisation et les dossiers réglementaires. Elle garantit la transparence, la traçabilité et la reproductibilité, qui sont indispensables tant pour répondre aux exigences réglementaires que pour assurer la traçabilité des données probantes générées par l'IA – des éléments clés mis en avant par les autorités réglementaires internationales et essentiels à la traçabilité des recherches basées sur l'IA
Exigences relatives à l'état de la technique en vertu des règlements EU MDR IVDR EU MDR
Tant dans le cadre EU MDR l'IVDR de l'UE, la définition de l'état de la technique (SOTA) est une exigence obligatoire pour l'évaluation clinique et l'évaluation des performances. Le SOTA correspond au niveau actuel et généralement admis des connaissances scientifiques, techniques et cliniques pertinentes pour le dispositif médical ou le dispositif de diagnostic in vitro.
Un protocole de recherche documentaire rigoureux est indispensable pour :
![]() Identifier les technologies de référence et les normes thérapeutiques
Identifier les technologies de référence et les normes thérapeutiques![]() Définir des profils de sécurité et de performance reconnus
Définir des profils de sécurité et de performance reconnus![]() Comparer l'appareil en question avec les alternatives actuelles
Comparer l'appareil en question avec les alternatives actuelles![]() Soutenir les activités liées aux programmes CER, PER, CEP, PEP, PMS et PMCF
Soutenir les activités liées aux programmes CER, PER, CEP, PEP, PMS et PMCF![]() Fournir des arguments solides pour justifier le rapport bénéfice/risque
Fournir des arguments solides pour justifier le rapport bénéfice/risque
 Identifier les technologies de référence et les normes thérapeutiques
Identifier les technologies de référence et les normes thérapeutiques Définir des profils de sécurité et de performance reconnus
Définir des profils de sécurité et de performance reconnus Comparer l'appareil en question avec les alternatives actuelles
Comparer l'appareil en question avec les alternatives actuelles Soutenir les activités liées aux programmes CER, PER, CEP, PEP, PMS et PMCF
Soutenir les activités liées aux programmes CER, PER, CEP, PEP, PMS et PMCF Fournir des arguments solides pour justifier le rapport bénéfice/risque
Fournir des arguments solides pour justifier le rapport bénéfice/risqueFreyr veille à ce que votre dossier de documentation démontre pleinement la conformité aux attentes SOTA, un facteur essentiel pour l'acceptation par l'organisme notifié.
Analyse EU MDR sur le règlement IVDR et le règlement EU MDR
La revue de la littérature EU IVDR/ EU MDR est un composant essentiel dans la gestion du cycle de vie d'un dispositif médical ou d'un DIV. Une stratégie systématique de recherche bibliographique EU IVDR/ EU MDR constitue la base des Clinical evaluation reports (CER), des rapports d'évaluation des performances (PER), de la surveillance post-commercialisation (PMS), des activités PMCF/PMPF ancrées dans la littérature basée sur des preuves pour les DIV/dispositifs médicaux, ce processus permet aux fabricants de soutenir une évaluation continue de la sécurité et des performances.
La revue EU MDR relative aux règlements IVDR et EU MDR comprend généralement :
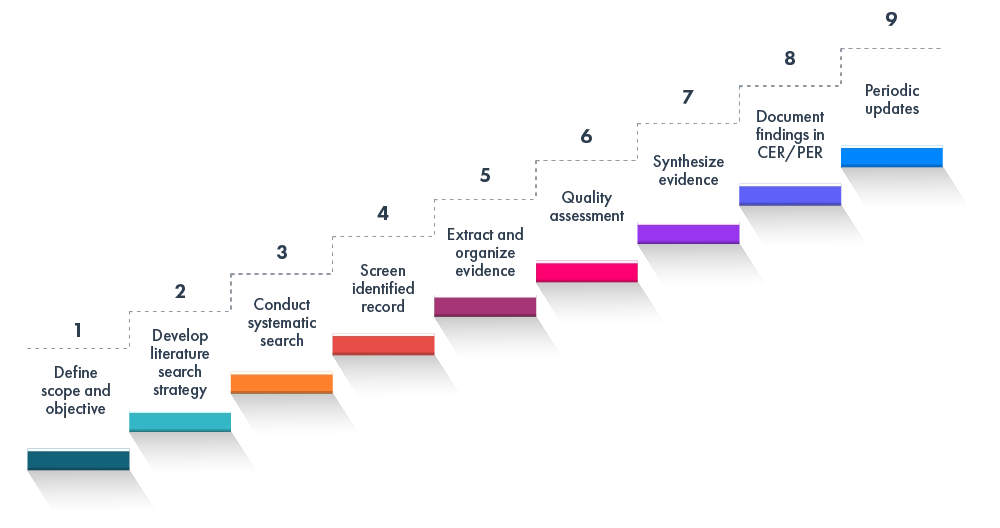
Ce cadre s'aligne sur les meilleures pratiques mondiales en matière d'évaluations cliniques et de performance ainsi que d'analyses documentaires.
Principales différences entre les exigences en matière de recherche documentaire de l'IVDR et du MDR
Bien que le règlement MDR et le règlement IVDR reposent tous deux sur une évaluation systématique des données probantes, leurs exigences diffèrent
MDR (dispositifs médicaux) met l'accent sur
![]() Évaluation clinique et données cliniques
Évaluation clinique et données cliniques![]() Allégations relatives à la sécurité et aux performances
Allégations relatives à la sécurité et aux performances![]() Collecte PMCF
Collecte PMCF![]() Justification du rapport bénéfice/risque
Justification du rapport bénéfice/risque![]() Alignement MEDDEV 2.7/1 Rév. 4
Alignement MEDDEV 2.7/1 Rév. 4
 Justification du rapport bénéfice/risque
Justification du rapport bénéfice/risqueL'IVDR (règlement sur les dispositifs médicaux de diagnostic in vitro) met l'accent sur
![]() Validité scientifique
Validité scientifique![]() Performances analytiques
Performances analytiques![]() Performances cliniques
Performances cliniques![]() Développement de PER et PEP
Développement de PER et PEP![]() Exigences en matière de preuves pour le PMPF
Exigences en matière de preuves pour le PMPF![]() Une reclassification plus stricte, nécessitant davantage de preuves à l'appui
Une reclassification plus stricte, nécessitant davantage de preuves à l'appui
 Validité scientifique
Validité scientifique Performances analytiques
Performances analytiques Développement de PER et PEP
Développement de PER et PEP Exigences en matière de preuves pour le PMPF
Exigences en matière de preuves pour le PMPF Une reclassification plus stricte, nécessitant davantage de preuves à l'appui
Une reclassification plus stricte, nécessitant davantage de preuves à l'appuiFreyr adapte ses stratégies de recherche documentaire, l'élaboration des études cliniques (CER/PER) et les protocoles relatifs aux services de surveillance post-commercialisation (PMCF), aux soins de santé post-commercialisation (PMCF) en fonction de la procédure réglementaire applicable au dispositif.
L'importance d'une équipe robuste de synthèse de la littérature scientifique
Pour s'y retrouver dans les exigences du MDR et de l'IVDR, il ne suffit pas de se contenter de simples recherches dans des bases de données. Une équipe spécialisée dans la synthèse de la littérature scientifique, dotée d'une expertise thérapeutique, garantit que votre analyse documentaire relative au MDR et à l'IVDR, votre protocole de recherche documentaire et votre documentation relative à l'évaluation clinique et des performances répondent aux critères de profondeur et de rigueur attendus par les autorités réglementaires.
Les experts de Freyr simplifient les processus complexes et transforment les données cliniques, de performance et scientifiques en preuves claires et solides qui renforcent les protocoles de recherche documentaire, les CER, les PER et les stratégies de PMS/PMPF.
Grâce à des méthodologies systématiques, des techniques de recherche avancées et des compétences en matière d'évaluation critique, notre équipe veille à ce que chaque analyse documentaire réponde aux exigences réglementaires internationales, tout en renforçant la qualité, la crédibilité et la préparation de votre dossier de données, ce qui confère à votre dispositif un avantage concurrentiel certain sur un marché en constante évolution.
Protocole de recherche EU MDR relatif au règlement IVDR et au règlement EU MDR
Un protocole de recherche EU MDR conforme aux règlements IVDR et EU MDR permet de structurer le processus, de réduire les biais des évaluateurs et de garantir une traçabilité totale lors des audits.
Le processus de recherche documentaire relatif à l'IVDR/MDR comprend :
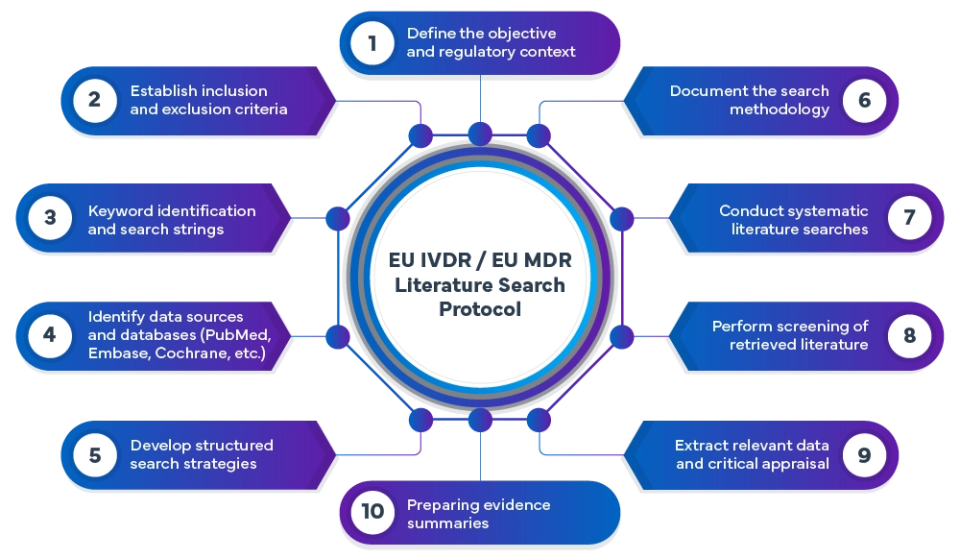
Chez Freyr, nous réalisons des analyses documentaires exhaustives conformes aux règlements MDR et IVDR en recourant à des méthodes de recherche avancées. Les publications issues de bases de données internationales sont systématiquement passées au crible et analysées afin d'identifier les données pertinentes qui étayent la sécurité, les performances et les avantages cliniques des dispositifs médicaux.
Protocole et analyse de la littérature relative aux dispositifs médicaux et aux produits de diagnostic in vitro (IVD)
- Recherche systématique, compilation et synthèse de la littérature scientifique
- Conception et mise en œuvre de protocoles de recherche documentaire conformes au règlement MDR/IVDR
- Définition des questions de recherche et des stratégies de recherche adaptées à l'outil.
- Identification des mots-clés, création de chaînes de recherche et sélection des bases de données (PubMed, Embase, Cochrane, etc.)
- Évaluation critique des données cliniques, des données de performance et des données scientifiques.
- Synthèse des éléments probants pour la documentation réglementaire.
- Rédaction de CER, CEP, PER et PEP
- Évaluation des lacunes dans les données issues de la documentation existante relative aux CER, CEP, PEP et PER.
- Utilisation de techniques de recherche avancées pour recenser la littérature mondiale pertinente

- Conformité garantie aux règlements MDR et IVDR
- Processus de recherche documentaire structuré, reproductible et justifiable
- Stratégies de collecte de preuves sur mesure et adaptées à chaque appareil
- Des experts hautement qualifiés dans les domaines clinique et réglementaire
- Capacité d'équipe évolutive ; stratégies de collecte de données sur mesure, adaptées à chaque appareil
- Contributions interfonctionnelles issues des domaines réglementaire, médical et clinique
- Prise en charge End-to-end de la recherche End-to-end , de l'analyse et de la documentation
- Renforce la crédibilité, la clarté et la qualité des dossiers réglementaires

Foire aux questions (FAQ)
01. Quel est l'objectif d'un protocole de recherche documentaire sur les dispositifs médicaux dans le cadre EU MDR?
Un protocole de recherche documentaire sur les dispositifs médicaux offre une approche structurée, systématique et transparente pour identifier, évaluer et documenter les données scientifiques relatives à un dispositif ou à ses produits de comparaison. Il garantit la reproductibilité, minimise les biais et permet aux autorités de réglementation de retracer la manière dont les données cliniques ou de performance ont été recueillies, évaluées et synthétisées. Dans le cadre EU MDR, un tel protocole soutient les évaluations de la sécurité, des performances et du rapport bénéfice/risque, garantit la conformité réglementaire et constitue la base d'une documentation de haute qualité et défendable tout au long du cycle de vie du dispositif.
02. En quoi les avancées technologiques influencent-elles l'évaluation clinique et l'évaluation des performances ?
Le « State-of-the-Art » (SOTA) correspond à l'état actuel des connaissances scientifiques et cliniques reconnues pour un type de dispositif. Il établit une référence en matière de sécurité, de performances et de résultats cliniques attendus. La définition du SOTA par le biais d'une analyse documentaire permet de replacer dans leur contexte les allégations relatives au dispositif, facilite le choix des dispositifs de comparaison et guide l'évaluation du rapport bénéfice/risque, la planification PMCF ainsi que la mise à jour des données tout au long du cycle de vie du dispositif.
03. En quoi une revue de la littérature MDR se distingue-t-elle d'une revue systématique traditionnelle ?
Une analyse documentaire MDR/IVDR se distingue d'une revue systématique traditionnelle en ce qu'elle est axée sur les autorités de réglementation et spécifiquement conçue pour faciliter la mise en conformité avec les exigences réglementaires. Alors que les revues systématiques traditionnelles visent à répondre à des questions de recherche scientifique et servent des objectifs purement académiques, une revue de la littérature MDR évalue les données cliniques et de performance afin de démontrer la sécurité, les performances et les profils bénéfice-risque des dispositifs. Elle suit une méthodologie structurée et traçable, avec des questions de recherche prédéfinies, des critères d'inclusion/d'exclusion et une évaluation critique, afin de produire une documentation défendable et prête pour un audit en vue des soumissions réglementaires.
04. À quelle fréquence les revues de la littérature concernant les dispositifs médicaux et les dispositifs de diagnostic in vitro doivent-elles être mises à jour ?
La fréquence des mises à jour dépend du niveau de risque du dispositif, de la dynamique du marché et de l'évolution des données disponibles. Les dispositifs à haut risque nécessitent généralement des mises à jour annuelles, tandis que d'autres peuvent suivre des intervalles définis. Les évaluations doivent également être révisées lorsque des signaux de sécurité importants, de nouvelles données cliniques, des avancées technologiques ou des modifications des recommandations apparaissent, afin de garantir l'exactitude des profils bénéfice-risque.
05. Quel rôle jouent les critères d'inclusion et d'exclusion dans les recherches documentaires relatives aux directives MDR et IVDR ?
Les critères d'inclusion et d'exclusion garantissent que seules les données pertinentes et de haute qualité sont sélectionnées. Ils renforcent l'objectivité, réduisent les biais des évaluateurs et assurent la cohérence du processus décisionnel. Dans le cadre des règlements MDR et IVDR, ces critères doivent être prédéfinis, justifiés et alignés sur les questions de recherche afin de garantir la traçabilité et la validité réglementaire tout au long du processus d'évaluation.
06. Pourquoi l'évaluation critique est-elle essentielle dans les revues de la littérature relatives aux règlements MDR et IVDR ?
L'évaluation critique porte sur la qualité méthodologique, la pertinence et la fiabilité des données incluses. Les cadres réglementaires MDR/IVDR accordent une importance particulière à cette évaluation, car les autorités de réglementation s'appuient sur des allégations de sécurité et de performance solidement étayées. Une évaluation rigoureuse permet de distinguer les données solides des études moins fiables et renforce les conclusions utilisées dans les analyses comparatives des preuves (CER), les évaluations de la performance (PER), les rapports de surveillance post-commercialisation (PMS) et les analyses bénéfice-risque.
07. En quoi les exigences en matière de recherche documentaire diffèrent-elles entre le règlement MDR et le règlement IVDR ?
Le règlement MDR met l'accent sur l'évaluation clinique, la justification du rapport bénéfice/risque et les performances cliniques, tandis que le règlement IVDR privilégie les performances analytiques, la validité scientifique et les performances cliniques en matière de précision diagnostique. Les stratégies documentaires doivent tenir compte de ces différences en adaptant les questions de recherche, les ensembles de données et les cadres d'évaluation aux différents parcours de démonstration de l'efficacité requis par chaque réglementation.
08. Quelles bases de données et sources d'information sont attendues dans le cadre des recherches documentaires menées conformément aux règlements MDR et IVDR ?
Les autorités de réglementation prévoient le recours à plusieurs bases de données scientifiques, telles que PubMed, Embase et Cochrane, complétées par des bases de données de vigilance, des registres d'essais cliniques, des recommandations et de la littérature grise pertinente. Le recours à des sources variées garantit une couverture exhaustive des informations cliniques, de performance et de sécurité nécessaires à une évaluation rigoureuse et à une surveillance continue.
09. Pourquoi Freyr est-il considéré comme un partenaire de premier plan dans le domaine de la recherche documentaire et des protocoles ?
Freyr est considéré comme un partenaire de premier plan grâce à sa parfaite maîtrise de la réglementation, à sa rigueur scientifique et à son respect constant des exigences en matière de données prévues par le RDM et l'IVDR. L'équipe applique les principes de l'analyse systématique, une méthodologie transparente et son expertise dans les domaines thérapeutiques pour produire des résultats solides et prêts à être soumis à un audit. L'approche de Freyr met l'accent sur la traçabilité, l'évaluation critique et l'analyse comparative par rapport à l'état de l'art, autant de facteurs clés appréciés par les organismes notifiés et les autorités réglementaires mondiales.







