
Aperçu de l'enregistrement des dispositifs médicaux en Israël
L'industrie israélienne des dispositifs médicaux connaît une croissance et une innovation soutenues, ce qui en fait un pôle pour les technologies de santé de pointe. L'enregistrement des dispositifs médicaux est crucial pour les entreprises qui entrent sur ce marché dynamique. Cet aperçu explore les aspects clés du processus d'enregistrement en Israël, offrant des perspectives sur le cadre réglementaire et les exigences pour placer les dispositifs médicaux innovants au premier plan du secteur de la santé en Israël.
Autorité de réglementation : La Division des dispositifs médicaux du ministère israélien de la Santé (AMAR).
Réglementation : Loi de 2012 sur les dispositifs/équipements médicaux
Voie réglementaire : Enregistrement de produit
Représentant local autorisé en Israël : Titulaire de l'enregistrement en Israël (IRH)
Exigence du SMQ : ISO 13485
Évaluation des données techniques : Le Département des dispositifs médicaux du Ministère de la Santé.
Validité de la licence : Cinq (05) ans
Format de soumission : Papier et électronique
Traduction : Documents traduits en hébreu
Classification des dispositifs
La loi sur les équipements médicaux et les réglementations relatives à l'enregistrement des équipements médicaux en Israël ne spécifient pas de système de classification des risques. Au lieu de cela, Israël aligne sa classification des dispositifs médicaux sur les normes internationales, en particulier celles définies par les pays du Groupe de travail sur l'harmonisation mondiale (GHTF). Alternativement, la classification des risques d'un dispositif dans un pays reconnu est adoptée pour l'enregistrement en Israël. Ce processus de classification prend généralement en compte l'utilisation prévue, le niveau de risque et d'autres facteurs susceptibles d'affecter la sécurité et l'efficacité des dispositifs médicaux.
Classes de dispositifs médicaux
| Classe | Risque |
|---|---|
| Classe I | Faible |
| Classe II | Faible à moyen |
| Classe III | Élevé |
Modifications proposées aux voies d'enregistrement
Les modifications proposées s'appliquent aux dispositifs de Classe I et de Classe II, tandis que le système d'enregistrement pour les dispositifs de Classe III reste inchangé.
- Les dispositifs de classe I peuvent être immédiatement enregistrés par auto-déclaration.
- Pour les dispositifs de classe II, bien que des déclarations et des documents techniques soient nécessaires, l'AMAR peut accélérer la procédure à quatorze (14) jours pour ceux jugés à risque faible à moyen. Cette mesure s'applique si le fabricant détient deux (02) autorisations délivrées par des pays reconnus et fournit six (06) mois de données de marché. Par ailleurs, pour les dispositifs de classe II ne disposant que de l'autorisationFDA (k)FDA US et de six (06) mois de données US , le délai de traitement par l'AMAR est ramené à soixante (60) jours.
Représentant autorisé local en Israël
Les entreprises de dispositifs médicaux basées en dehors d'Israël doivent désigner un Titulaire d'enregistrement israélien (IRH) pour faciliter l'enregistrement de leurs produits destinés à la vente dans le pays. L'IRH agit en tant que représentant local du fabricant et est chargé d'assurer la liaison avec le ministère de la Santé pour garantir le respect des réglementations locales. De plus, un IRH est responsable de l'établissement et du maintien d'une présence commerciale en Israël, ainsi que de l'obtention et du maintien d'une licence commerciale valide.
Enregistrement des dispositifs médicaux
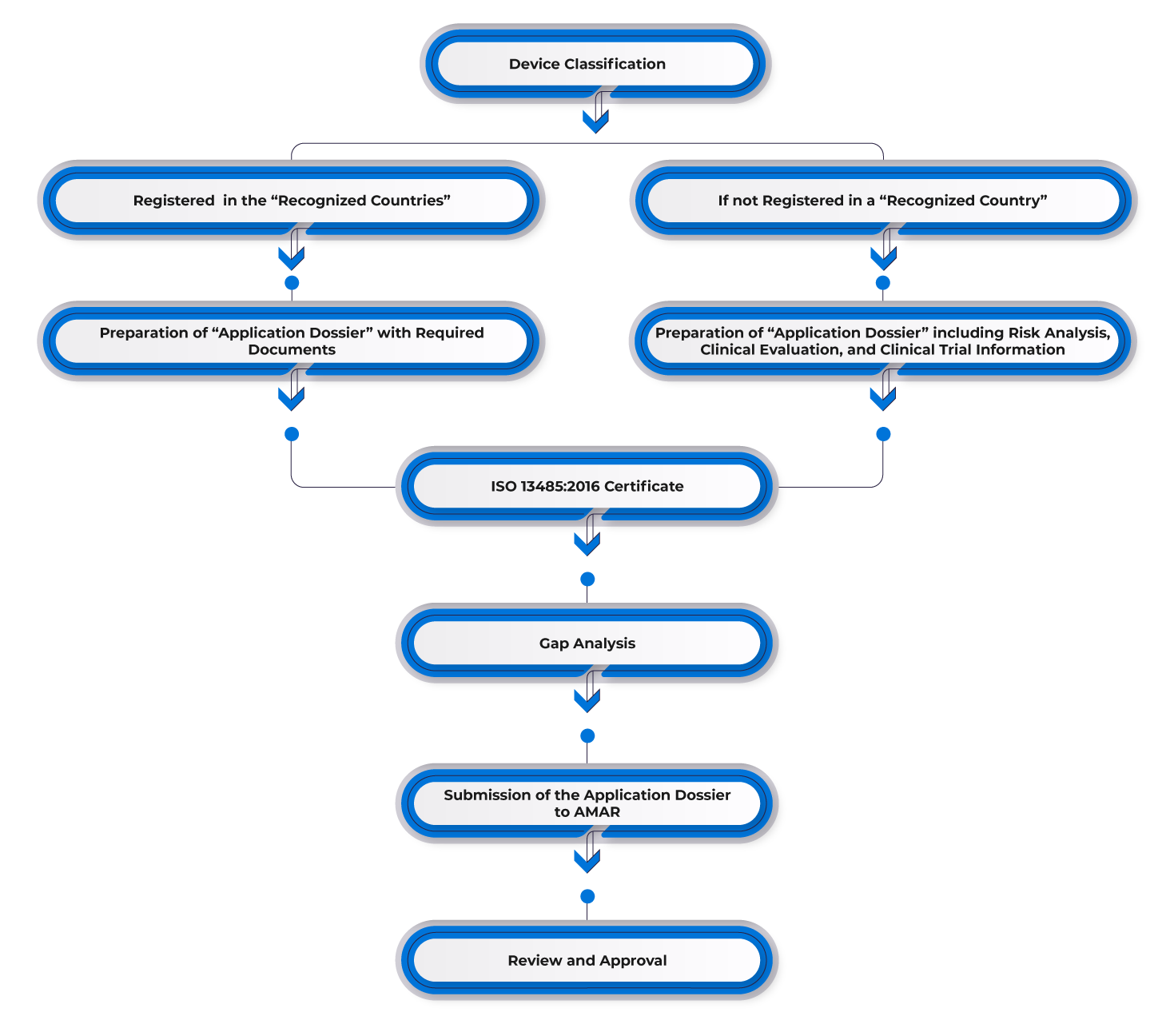
Pour enregistrer un dispositif médical en Israël, les fabricants doivent obtenir une approbation préalable sur l'un des marchés de référence tels que les US, l'Europe, l'Australie, le Canada ou d'autres marchés majeurs. Les fabricants ayant déjà des approbations dans l'un des pays de référence peuvent utiliser cette approbation pour le marché israélien et désigner un représentant local. Par la suite, ils doivent soumettre la documentation requise, notamment :
- FDA (k)/Lettre d'autorisation préalable à la mise sur le marché/Marquage CE.
- Certificat destiné à un gouvernement étranger (CFG)/Certificat de Libre Vente (CFS).
- ISO 13485 une autre certification relative aux bonnes pratiques de fabrication (BPF) reconnue.
- Validation et certification de l'Institut des normes d'Israël (Si nécessaire).
Flux de processus
Gestion du cycle de vie des dispositifs après approbation
Freyr offre un soutien complet aux fabricants étrangers pour la gestion de l'ensemble du cycle de vie des dispositifs médicaux en Israël, y compris les activités post-approbation :
- Gestion des modifications post-approbation, traitant des modifications aux approbations existantes de dispositifs médicaux, telles que l'ajout de nouvelles variantes, d'accessoires et d'indications d'utilisation.
- Maintien de la certification ISO 13485:2016 et de la certification CE.
- Renouvellement des licences.
- Agir en tant qu'intermédiaire entre l'Organisme Notifié (ON) et le fabricant.
Gérer les subtilités des organismes d'autorisation et se conformer à plusieurs ensembles de réglementations pour les approbations de dispositifs peut être difficile. Obtenir des approbations de divers pays du GHTF et respecter les réglementations spécifiques à chaque État nécessitent une connaissance réglementaire approfondie. Pour les nouveaux entrants sur le marché confrontés à ces complexités sans partenaire réglementaire établi, Freyr propose des services réglementaires End-to-End, simplifiant le processus d'approbation des dispositifs médicaux en Israël.
Expertise en enregistrement des dispositifs médicaux en Égypte
- Classification des dispositifs médicaux en Israël.
- Titulaire de l'enregistrement en Israël (IRH).
- Enregistrement des dispositifs en Israël.
- Consultation en gestion des risques ISO 14971:2019.
- Conformité à la norme ISO 13485:2016.
- Examen, compilation et soumission du dossier de conception.
- Enregistrement des dispositifs médicaux via le système d'enregistrement en ligne.
- Rapport sur la stratégie réglementaire des dispositifs médicaux.
- Soutien aux essais - Biocompatibilité, Sécurité électrique, Mécanique et Performance.
- Soutien à la conformité du labelling.
- Soutien GMP.
- Soutien à la surveillance après commercialisation (PMS).
