
Aperçu de la description des travaux (SOW) pour les demandes 510(k) concernant les dispositifs médicaux actifs et non actifs
Chez Freyr, notre équipe d'experts compile et synthétise avec rigueur les informations les plus récentes indispensables à vos demandes 510(k), qu'il s'agisse de dispositifs médicaux actifs ou non actifs. Vous disposez ainsi des connaissances nécessaires pour naviguer en toute confiance dans le cadre réglementaire. Qu'il s'agisse de clarifier les différences entre les dispositifs actifs et non actifs ou d'approfondir les subtilités de la demande 510(k), nous avons constitué une vaste base de ressources qui vous servira de référence principale. Lancez-vous dans l'aventure pour maîtriser les demandes 510(k) relatives aux dispositifs médicaux actifs et aux dispositifs médicaux non actifs grâce à notre guide complet.
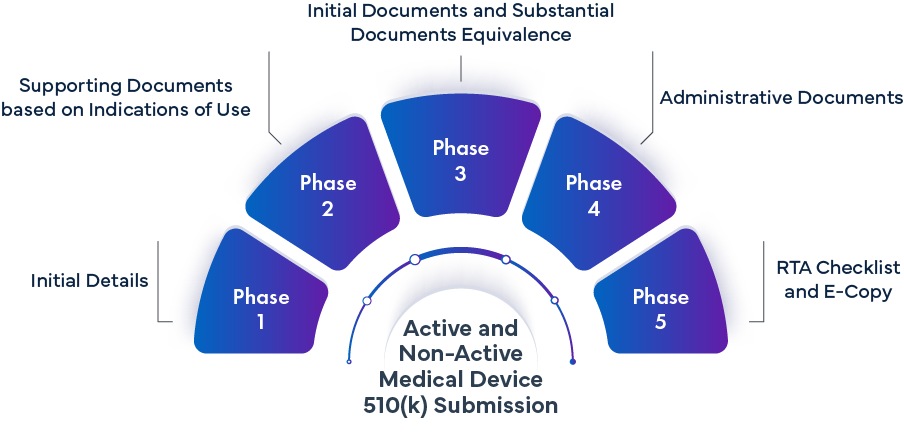
Phase -1 Détails initiaux | ||
|---|---|---|
Exigences | Portée du demandeur 510(k) | Champ d'action de Freyr |
| Utilisation prévue |
|
|
| Déclaration des indications d'utilisation (Form3881) |
|
|
| Description du dispositif |
|
|
| Normes et lignes directrices |
|
|
| Dispositif de référence |
|
|
| Résumé de la procédure 510(k) |
|
|
Phase 2 : Documentation justificative basée sur les indications d'utilisation | |||
|---|---|---|---|
Exigences documentaires | Portée du demandeur 510(k) | Champ d'action de Freyr | |
| 2.1 | Plan du dispositif | Soumettre le fichier de dessins du dispositif afin d'assurer une représentation précise de la conception du dispositif. | Initier une demande formelle pour le plan d'un dispositif actif. Examiner minutieusement et documenter avec précision les informations nécessaires pour la soumission 510(k). |
| 2.2 | Conception et développement du dispositif | Soumettre le dossier de conception et de développement du dispositif actif, englobant toutes les informations et la documentation pertinentes. | Soumettre une demande concernant la conception et le développement d'un dispositif actif. Examiner minutieusement et documenter avec précision toutes les informations nécessaires en préparation de la soumission 510(k). |
| 2.3 | Fiche de données de sécurité des matériaux | Fournir les Fiches de Données de Sécurité (MSDS) pour les composants essentiels du dispositif actif, afin de garantir des informations complètes concernant leur sécurité et leur composition. | Envoyer une demande de fiche de données de sécurité des composants essentiels du dispositif actif. Examiner minutieusement et documenter avec précision toutes les informations nécessaires en préparation de la soumission 510(k). |
| 2.4 | Organigramme de fabrication | Fournir un organigramme de fabrication détaillant le processus de production du dispositif actif, offrant une représentation visuelle des étapes de fabrication et de leur séquence. | Soumettre une demande pour la fiche de données de sécurité (MSDS) des composants essentiels pour le dispositif actif. Examiner minutieusement et documenter avec précision toutes les informations nécessaires en préparation de la soumission 510(k). |
| 2.5 | Description du dispositif | Fournir des détails complets, notamment : o Un aperçu de l'appareil o Fonctions et modes de fonctionnement o Schémas fonctionnels o Photographies, câbles et accessoires pertinents o Interopérabilité du dispositif. o Description de l'alimentation électrique | Soumettre une demande d'informations détaillées sur le dispositif. Examiner minutieusement et documenter avec précision toutes les informations nécessaires en préparation de la soumission 510(k). |
| 2.6 | Proposition Labelling | Fournir les Instructions d'Utilisation (IFU), le Manuel d'Utilisation et tout matériel promotionnel associé pour le dispositif actif. | Soumettre une demande pour les instructions d'utilisation (IFU), le manuel d'utilisation et tout matériel promotionnel, le cas échéant. Examiner les instructions d'utilisation, le manuel d'utilisation et le matériel promotionnel fournis par le demandeur. Documenter la notice d'utilisation (IFU), le manuel d'utilisation et le matériel promotionnel aux fins de la soumission 510(k). |
| 2.7 | Emballage et transport | Fournir les plans et rapports d'étude pour la validation de l'emballage et du transport. | Soumettre une demande concernant le plan d'étude et les rapports de validation de l'emballage et du transport. Examiner les plans et rapports d'étude pour la validation de l'emballage et du transport et fournir les corrections ou commentaires nécessaires. |
| 2.8 | Stérilisation (Si la stérilité est applicable) | Fournir les plans et rapports d'étude pour la validation de la stérilisation. | Soumettre une demande concernant le plan d'étude et les rapports de validation de la stérilisation. Examiner minutieusement et documenter avec précision toutes les informations nécessaires en préparation de la soumission 510(k). |
| 2.9 | Tests de performance _ Banc | Initier une demande officielle pour les plans et rapports des études de performance sur banc d'essai, décrivant les exigences et objectifs spécifiques à atteindre | Soumettre une demande concernant les plans et rapports d'étude sur banc du dispositif actif pour les tests de performance. Examiner minutieusement et documenter avec précision toutes les informations nécessaires en préparation de la soumission 510(k). |
Documents justificatifs relatifs à la compatibilité électromagnétique et à la sécurité électrique | |||
| 2.10 | Caractéristiques des dispositifs liées à la CEM et environnements d'utilisation prévue | Fournir des détails sur les caractéristiques du dispositif liées à la CEM et les environnements d'utilisation prévue, notamment : o Un aperçu de l'appareil. o Fonctions et modes de fonctionnement. o Schémas fonctionnels. o Photographies, câbles et accessoires pertinents. o Interopérabilité du dispositif. o Description de l'alimentation électrique, y compris la faisabilité d'utiliser le dispositif médical alimenté en interne pendant la charge. o Environnements dans lesquels le dispositif médical est destiné à être utilisé. o Description de toute technologie sans fil (le cas échéant) pour des considérations supplémentaires concernant les dispositifs médicaux dotés de capacités sans fil. o Description de tout émetteur RF interne dans le dispositif médical susceptible de provoquer des perturbations électromagnétiques. o Aborder les émetteurs électromagnétiques (EM) courants ainsi que les émetteurs médicaux uniques.
| Soumettre une demande d'informations concernant les caractéristiques du dispositif liées à la CEM et les environnements d'utilisation prévus. Examiner minutieusement et documenter avec précision toutes les informations nécessaires en préparation de la soumission 510(k). |
| 2.11 | Évaluation des risques | Fournir un plan de gestion des risques qui comprend une évaluation des risques mettant en évidence une atténuation efficace des risques, ainsi qu'un rapport complet de gestion des risques englobant tous les éléments de risque. Fournir le document révisé avec les corrections et améliorations suggérées | Soumettre une demande concernant le dossier de gestion des risques et demander la documentation du plan et du rapport de gestion des risques, incluant l'identification des dangers liés aux risques, l'évaluation des risques et la démonstration d'une atténuation appropriée des risques. Le rapport de gestion des risques doit couvrir tous les éléments de risque, de préférence avec des sections distinctes pour plus de clarté. Fournir un modèle de plan de gestion des risques et de rapport de gestion des risques qui couvre tous les risques liés au dispositif, sur demande du demandeur. Examiner les données du dossier de gestion des risques, y compris le Plan et le Rapport fournis par le demandeur, et formuler des suggestions de corrections nécessaires afin de garantir une documentation complète pour la soumission 510(k). Examiner minutieusement et documenter avec précision toutes les informations nécessaires en préparation de la soumission 510(k). |
| 2.12 | Norme consensuelle | Veuillez fournir une confirmation des normes consensuelles applicables et une explication de tout écart par rapport aux normes FDA. | Soumettre une demande concernant les normes consensuelles applicables liées à la CEM et à la sécurité électrique pour le dispositif actif. Documenter les normes consensuelles confirmées pour le dispositif actif aux fins de la soumission 510(k). |
| 2.13 | Critères de réussite/échec pour la performance essentielle et l'immunité | Veuillez soumettre le plan d'étude et les rapports relatifs aux essais de performances essentielles et d'immunité réalisés sur le dispositif en service, conformément aux normes FDA. | Soumettez une demande concernant le protocole d'étude et les rapports relatifs aux essais de performance et d'immunité essentiels réalisés sur le dispositif en service, conformément aux normes FDA. Examiner minutieusement et documenter avec précision toutes les informations nécessaires en préparation de la soumission 510(k). |
| 2.14 | Configuration et fonctionnalités des dispositifs médicaux testées | Fournir la configuration du dispositif médical et les fonctions testées pour le dispositif actif, en incluant les détails suivants : o Fournir une description complète du dispositif médical testé, incluant des informations détaillées sur sa configuration, ses fonctions, ses modes et les réglages spécifiques qui ont été testés. o La description du dispositif testé doit inclure le nom du dispositif médical, son numéro de modèle, et indiquer si le dispositif est le dispositif médical prêt pour la production finale actuellement en cours d'examen. | Soumettre une demande concernant la configuration et les fonctions de test du dispositif médical actif. Examiner minutieusement et documenter avec précision toutes les informations nécessaires en préparation de la soumission 510(k). |
| 2.15 | Résultats des tests de compatibilité électromagnétique (CEM) | Fournir le plan d'étude et le rapport relatifs aux essais de compatibilité électromagnétique (CEM), conformément à la norme consensuelle FDA et recommandée pour le dispositif actif. | Lancer une demande officielle concernant le plan d'étude et le rapport d'essais CEM, conformément à la norme consensuelle FDA et recommandée pour les dispositifs actifs. Examiner minutieusement et documenter avec précision toutes les informations nécessaires en préparation de la soumission 510(k). |
Phase 3 – Documents initiaux et documents d'équivalence substantielle | |||
|---|---|---|---|
Exigences documentaires | Portée du demandeur 510(k) | Champ d'action de Freyr | |
| 3.1 | Fiche de couverture pour la demande d'examen préalable à la mise sur le marché auprès du CDRH (FDA 3514) | - | Remplissez le FDA 3514 FDA en utilisant les informations fournies par le demandeur |
| 3.2 | Résumé et certification de classe III | - | Cette étape n'est pas nécessaire si les études cliniques ne sont pas exigées. |
| 3.3 | Certification financière ou déclaration de divulgation | - | Cette étape n'est pas nécessaire si les études cliniques ne sont pas exigées. |
| 3.4 | Résumé analytique | - | Élaborer un modèle et préparer méticuleusement le document. Fournir des justifications pour toute divergence observée entre le dispositif proposé et le dispositif de référence. Si une étude comparative entre le dispositif proposé et le dispositif de référence est choisie, créer un modèle et préparer le document correspondant. |
| 3.5 | Discussion sur l'équivalence substantielle | - | Élaborer un modèle et préparer méticuleusement le document. Si une étude comparative entre le dispositif proposé et le dispositif de référence est choisie, créer un modèle et préparer le document correspondant. |
Phase 4 – Documents administratifs | |||
|---|---|---|---|
Exigences documentaires | Portée du demandeur 510(k) | Champ d'action de Freyr | |
| 4.1 | Lettre de présentation 510(k) | Signez le document imprimé sur papier à en-tête de l'entreprise et organisez l'envoi d'une copie papier par courrier au bureau US. Fournir une copie numérique de la lettre de présentation 510(k) signée pour inclusion dans la documentation 510(k). | Préparer un modèle complet incluant tous les détails nécessaires pour la lettre d'accompagnement et le fournir au demandeur. Demander au demandeur d'utiliser son papier à en-tête officiel et s'assurer que la lettre d'accompagnement est signée par une personne autorisée |
| 4.2 | Déclaration d'exactitude et de véracité | Assurez que le document est signé par la personne de contact désignée au sein de l'entreprise et fourni en conséquence. | Développer un modèle complet contenant tout le contenu nécessaire à inclure dans le document de soumission. |
| 4.3 | Déclarations de conformité et rapport de synthèse | Assurez que le document est signé par la personne de contact désignée au sein de l'entreprise et fourni en conséquence. | Développer un modèle complet pour lister et préparer systématiquement les documents requis. |
| 4.4 | MDFUSC (FDA 3601FDA ) | Effectuez le paiement requis auprès de la FDA le dépôt officiel du dossier 510(k) | Générer une page de garde des frais d'utilisateur et un numéro d'identification personnel (NIP) unique spécifiquement pour le dépôt de dispositifs médicaux. |
Phase 5 – Liste de contrôle RTA et copie électronique | |||
|---|---|---|---|
Exigences documentaires | Portée du demandeur 510(k) | Champ d'action de Freyr | |
| 5.1 | Liste de contrôle RTA | Approbation de la vérification de la liste de contrôle RTA (Prêt à accepter), indiquant que toutes les exigences ont été remplies avec succès | Élaborer un modèle de liste de contrôle RTA personnalisé et adapté au type spécifique de soumission. Veuillez remplir la liste de contrôle en complétant minutieusement tous les champs obligatoires et en vous assurant que les documents mentionnés ont bien été transmis à la FDA communiqués au demandeur. |
| 5.2 | Copie électronique | Approbation de la documentation contenue dans le dossier de soumission finale, attestant de sa conformité à toutes les exigences et normes nécessaires. | Organisez les sections du dossier de demande conformément aux FDA et transmettez-les sans délai au demandeur. Convertir le dossier de soumission en une copie électronique pour un accès et un examen facilités. Soumettez la copie électronique de la soumission à l'agent US désigné. |
Enregistrement des dispositifs médicaux
- StratégieFDA globale auprès deFDA US
- Identification du dispositif de référence
- Établissement de l'équivalence substantielle avec un dispositif de référence
- Analyse des écarts en matière deFDA US
- Compilation des 21 sections du dossier technique 510(k).
- Publication et création d'eCopy
- Validation et soumission de l'eCopy
- Services de liaison pour l'approbation des dispositifs
- Traitement de la réponse RTA et des lacunes.
- Services de consultation pour remédier aux lacunes
- Enregistrement des dispositifs et maintenance de la base de données FURLS

- Nous avons géré de nombreux enregistrements 510(k) pour des catégories de dispositifs diversifiées.
- Équipe d'experts spécialisée dans la constitution des dossiers 510(k) conformément aux exigences de la notificationFDA (510(k))FDA US
- Soutien supplémentaire pour gérer les demandes 510(k).
- Conseils pour choisir le type de demande 510(k) approprié, conformément aux exigences deFDA US en matière de soumissionFDA (k) pour ce dispositif
- Soumission dans les délais des livrables
- À jour avec lesFDA modifications apportées parFDA US
