


Aperçu de l'enregistrement des dispositifs médicaux en Turquie
Le marché turc des dispositifs médicaux a connu une croissance significative et constante au cours de la dernière décennie. À partir de 2021, l'enregistrement des dispositifs médicaux en Turquie exige de se conformer au Règlement de l'UE sur les dispositifs médicaux (MDR) 2017/745 et au Règlement sur les dispositifs médicaux de diagnostic in vitro (IVDR) 2017/746. Cela a renforcé le commerce international, conduisant plusieurs entreprises mondiales à lancer leurs dispositifs médicaux dans le pays.
![]()
Autorité réglementaire : Agence turque des médicaments et des dispositifs médicaux (TITCK)![]()
Réglementation : Règlement sur les dispositifs médicaux (MDR) 2017/745, Règlement sur les dispositifs médicaux de diagnostic in vitro 2017/746![]()
Voie réglementaire : Le marquage CE est obligatoire, suivi d'un enregistrement/d'une notification dans le Système de Suivi des Produits (UTS)![]()
Représentant local en Turquie![]()
Exigence SMQ : ISO 13485:2016![]()
Évaluation des données techniques : Organisme notifié pour le marquage CE![]()
Validité de la licence : Illimité![]()
Format de soumission : Papier![]()
Traduction : Documents traduits en turc
Classification des dispositifs
La Turquie suit la même classification des dispositifs médicaux que celle définie dans l'EU MDR et l'IVDR. Déterminer la classification des dispositifs peut être difficile, c'est pourquoi le soutien d'un consultant réglementaire expérimenté est essentiel.
Classes de dispositifs médicaux
| Classe | Risque |
|---|---|
| Classe I | Faible |
| Classe IIa | Modéré |
| Classe IIb | Modéré à élevé |
| Classe III | Élevé |
Classes de dispositifs médicaux de diagnostic in vitro
| Classe | Risque |
|---|---|
| Classe A | Faible |
| Classe B | Modéré |
| Classe C | Modéré à élevé |
| Classe D | Élevé |
Enregistrement des dispositifs médicaux
Le marquage CE est une conformité requise par les fabricants pour commercialiser leur dispositif sur le marché turc. Le marquage CE est délivré suite à une évaluation de la conformité réalisée par l'organisme notifié. Désormais, la Turquie est autorisée à désigner des organismes notifiés conformément à l'EU MDR et à l'IVDR.
Les entreprises sont tenues de s'enregistrer dans le Système central d'enregistrement (MERSIS) et d'enregistrer le dispositif dans le Système de suivi des produits (UTS).
Flux de processus
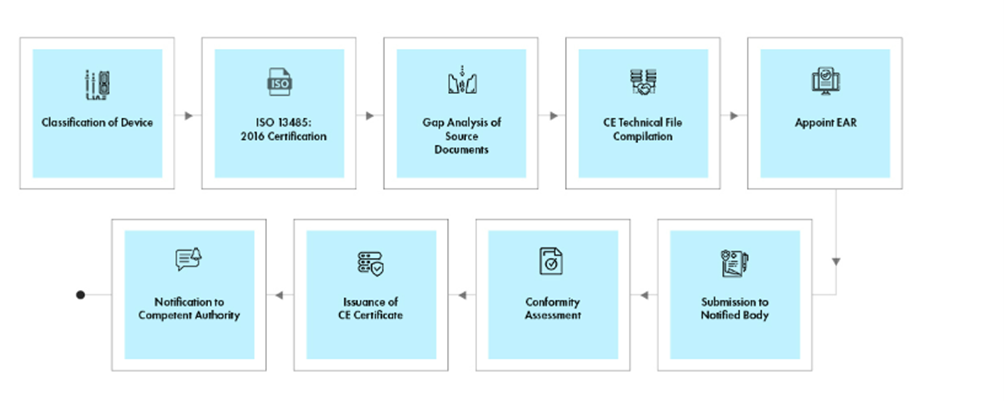
Gestion du cycle de vie des dispositifs après approbation
Freyr accompagne les fabricants étrangers dans la gestion End-to-end du cycle de vie des dispositifs médicaux, y compris les activités post-approbation, telles que :
- Gestion des modifications post-approbation – modifications aux approbations existantes de dispositifs médicaux, telles que l'ajout de nouvelles variantes, d'accessoires, et l'ajout de nouvelles indications d'utilisation, entre autres
- Maintien de la certification ISO 13485:2016 et de la certification CE
- Renouvellement des licences
- Assurer la liaison entre l'organisme notifié et le fabricant
Avec diverses autorités d'autorisation impliquées, les fabricants étrangers doivent se conformer à plusieurs ensembles de réglementations dans chaque processus individuel pour les approbations de dispositifs. L'obtention du marquage CE et le respect des réglementations spécifiques à chaque État nécessitent une connaissance réglementaire approfondie. Parfois, sans un partenaire réglementaire éprouvé, naviguer à travers toutes les exigences relatives aux dispositifs peut être difficile pour les nouveaux entrants sur le marché. Pour aider les fabricants, Freyr fournit des services réglementaires End-to-End pour accélérer les approbations des dispositifs médicaux.
L'expertise de Freyr
- Classification européenne des dispositifs médicaux
- Support de Représentant autorisé européen (EAR)
- Enregistrement des dispositifs et notification de produits en Turquie
- Consultation en gestion des risques ISO 14971:2019
- Conformité à ISO 13485:2016
- Examen, compilation et soumission du dossier technique CE/dossier de conception
- Soutien à la transition EU MDR
- Soutien à la transition IVDR de l'UE
- Clinical Evaluation Reports (CER) pour les dispositifs médicaux
- Rapports d'évaluation des performances (PER) pour les dispositifs médicaux de diagnostic in vitro
- Notification/Enregistrement des dispositifs médicaux via le système d'enregistrement en ligne
- Rapport sur la stratégie réglementaire des dispositifs médicaux
- Soutien aux essais - biocompatibilité, sécurité électrique, mécanique et performance
- Soutien à la conformité du labelling
- Soutien GMP
- Soutien à la surveillance après commercialisation
