Qu'est-ce que l'étiquetage réglementaire ?
Dernière mise à jour le : août 2024
L'étiquetage réglementaire pharmaceutique implique la création, l'examen et la gestion de documents essentiels qui communiquent des informations produit essentielles aux parties prenantes, assurant la conformité aux normes réglementaires mondiales. Les composants principaux incluent le CDS et le CCDS, dérivés de sources telles que les brochures pour investigateurs et les données post-commercialisation. Ce processus est essentiel pour transmettre les informations de sécurité et d'efficacité sur les étiquettes spécifiques à chaque pays et pour s'aligner sur les exigences des autorités de santé (HA).
Axé sur l'harmonisation mondiale, l'étiquetage réglementaire pharmaceutique répond aux exigences réglementaires en évolution, englobant les nouvelles autorisations de produits, les soumissions aux autorités sanitaires (HA), les post-approbations et la gestion du cycle de vie. La précision et le respect des directives en évolution sont cruciaux pour un étiquetage réglementaire pharmaceutique réussi, influençant l'autorisation de mise sur le marché d'un produit, son profil de sécurité et sa viabilité réglementaire globale.

Freyr, un leader des services de labelling réglementaire des médicaments End-to-End, dispose d'une équipe dédiée de plus de 180 experts mondiaux en labelling, excellant dans la rédaction de documents cruciaux tels que les brochures pour investigateurs (IB), les fiches de données de base de développement et les informations de sécurité de base de développement. L'exploitation de l'intelligence artificielle améliore la précision et accélère la mise en œuvre et l'examen des fiches de données. Avec un modèle CCDS rationalisé et des processus axés sur la précision, les services complets de Freyr répondent efficacement aux besoins dynamiques de l'industrie pharmaceutique, offrant un soutien inégalé pour la Conformité du labelling et le succès réglementaire.
Pourquoi l'étiquetage réglementaire est-il important dans l'industrie pharmaceutique ?
- Assurer la sécurité des patients et la communication des informations : L'étiquetage réglementaire est crucial pour la sécurité des patients. Les étiquettes fournissent des informations essentielles sur l'utilisation des médicaments, les dosages, les effets secondaires et les contre-indications. Les patients, les médecins prescripteurs, les professionnels de la santé et les soignants se fient à ces étiquettes pour prendre des décisions éclairées. Un étiquetage clair et précis réduit le risque d'erreurs médicamenteuses, d'événements indésirables et de mauvaise utilisation.
Il garantit que les patients reçoivent le bon traitement et en comprennent l'utilisation appropriée. De plus, les autorités réglementaires exigent que chaque produit pharmaceutique sur le marché dispose d'un étiquetage pour communiquer efficacement les informations sur le traitement. - Conformité et atténuation des risques : Le respect de la réglementation en matière d'étiquetage n'est pas une simple formalité, mais une obligation légale. Les organismes de réglementation tels que la US and Drug Administration (FDA) US , l'Agence européenne des médicaments (EMA) et d'autres exigent un étiquetage précis et exhaustif. Tout manquement à cette obligation peut entraîner des amendes réglementaires, nuire à la réputation de la marque, voire entraîner l'arrêt temporaire des chaînes de production. Les entreprises pharmaceutiques doivent démontrer que leurs processus, méthodes, tests et équipements d'étiquetage sont capables de produire de manière constante des produits sûrs et efficaces. Un étiquetage correctement validé atténue les risques et garantit le respect des bonnes pratiques de fabrication (BPF).
- Accès au marché et harmonisation mondiale: Des étiquettes bien structurées facilitent l'accès au marché mondial. Un labelling cohérent entre les régions rationalise les processus, réduit les redondances et s'aligne sur les normes harmonisées. À mesure que les régulateurs internationaux adoptent les exigences de validation BPF, y compris la sérialisation, les chaînes d'approvisionnement pharmaceutiques sont confrontées à une complexité croissante. Les entreprises qui privilégient la conformité du labelling établissent la confiance, améliorent l'acceptation du marché et se positionnent pour réussir dans un paysage concurrentiel.
Qu'est-ce que le processus d'approbation de l'étiquetage ?
Le processus d'approbation de l'étiquetage dans l'industrie pharmaceutique implique plusieurs étapes pour garantir que toutes les informations relatives au médicament sont exactes, conformes et claires pour les professionnels de la santé et les patients. Il commence par la rédaction du contenu de l'étiquette, qui comprend des détails sur la posologie, l'administration, la sécurité et les avertissements. Les équipes réglementaires et médicales examinent ensuite ce projet en interne pour s'assurer qu'il est conforme aux normes réglementaires locales et internationales. Une fois finalisée, l'étiquette est soumise aux autorités sanitaires pour approbation, où elle subit un examen rigoureux pour vérifier sa conformité aux exigences de sécurité et d'efficacité. Ce n'est qu'après avoir reçu l'approbation officielle que l'étiquette peut être utilisée pour la commercialisation du médicament.
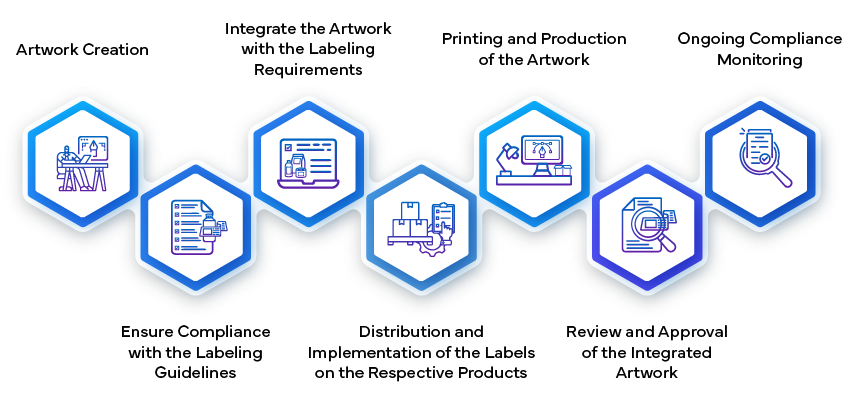
- Création et soumission de l'étiquetage:
- Collecte initiale des données : Les entreprises pharmaceutiques compilent toutes les données pertinentes pour l'étiquetage des médicaments. Cela inclut des informations sur l'efficacité, la sécurité, les dosages, les indications, les contre-indications et les risques potentiels. L'étiquette sert d'outil de communication essentiel pour les professionnels de la santé et les patients.
- Soumission aux autorités réglementaires: L'entreprise transmet les données relatives à l'étiquetage aux organismes de réglementation, tels que la US and Drug Administration (FDA) US ou l'Agence européenne des médicaments (EMA). Ces organismes évaluent les données afin de s'assurer que le médicament présente des bénéfices qui l'emportent sur ses risques connus et potentiels pour la population ciblée.
- Analyse contextuelle : Les évaluateurs analysent la maladie ou l'affection cible pour laquelle le médicament est destiné. Ils prennent en compte le paysage thérapeutique existant, en évaluant les risques du médicament par rapport à ses bénéfices. Par exemple, un médicament traitant une maladie mortelle sans thérapie alternative peut être approuvé même si les risques seraient inacceptables pour une affection non mortelle.
- Évaluation des données cliniques: FDA évaluent les informations relatives aux bénéfices et aux risques cliniques fournies par le laboratoire pharmaceutique. Ils tiennent compte des incertitudes pouvant découler de données imparfaites ou incomplètes. En règle générale, l'agence attend des résultats issus d'essais cliniques bien conçus pour valider l'efficacité et la sécurité du médicament.
- Établissement de l'Artwork:
- Une fois le contenu de l'élément labelling approuvé, l'étape suivante consiste à créer l'Artwork de l'élément labelling. Cela implique la conception des éléments visuels, de la mise en page, des polices et des graphiques. L'Artwork doit être conforme aux directives réglementaires et représenter fidèlement les informations figurant sur l'élément labelling.
- L'Artwork de l'étiquette fait l'objet d'examens internes au sein de l'entreprise pharmaceutique pour garantir la cohérence et la conformité. Il comprend des détails tels que les instructions de dosage, les avertissements, les conditions de stockage et les coordonnées.
- L'Artwork final est soumis aux autorités réglementaires pour approbation. Cette étape garantit que la présentation visuelle de l'étiquette respecte les normes de qualité et communique efficacement les informations essentielles aux utilisateurs.
- Fabrication et mise en œuvre:
- Une fois approuvé, l'Artwork de l'élément labelling devient une partie intégrante de l'emballage du médicament. Les fabricants s'assurent que les éléments labelling sont correctement apposés sur chaque unité de produit (par exemple, flacons, plaquettes thermoformées, fioles).
- Les procédures de contrôle qualité vérifient que les étiquettes répondent aux spécifications, notamment en ce qui concerne l'exactitude du contenu, la lisibilité et le respect des directives de conception.
- L'étiquette sert de pont entre l'entreprise pharmaceutique, les agences de réglementation, les professionnels de la santé et les patients. Elle joue un rôle vital pour garantir une utilisation sûre et efficace des médicaments tout au long du cycle de vie du produit.
Quels sont les défis courants en matière d'étiquetage réglementaire ?
Les défis courants en matière d'étiquetage réglementaire incluent le suivi des exigences réglementaires en constante évolution, la gestion de l'étiquetage multilingue et l'assurance de la cohérence à travers des gammes de produits diverses. La nature dynamique des normes réglementaires pour l'étiquetage pharmaceutique exige une vigilance constante pour rester informé des dernières exigences. S'adapter aux réglementations changeantes et mettre en œuvre rapidement les mises à jour nécessaires du contenu et du format de l'étiquetage est essentiel pour maintenir la conformité et assurer la sécurité des patients.
| Défi | Description |
|---|---|
| Exigences réglementaires en évolution. | Faire face aux réglementations et aux lignes directrices en constante évolution, nécessitant des mises à jour constantes de la documentation d'étiquetage. |
| Harmonisation mondiale | Assurer la cohérence des informations sur les produits dans diverses régions, en s'alignant sur les exigences variées des différentes autorités sanitaires. |
| Intégration des données après commercialisation | Gérer l'intégration des données de sécurité et d'efficacité post-commercialisation dans l'étiquetage, en maintenant l'exactitude et la pertinence. |
| Conformité aux normes d'étiquetage locales | Respecter les normes d'étiquetage spécifiques de chaque pays, en tenant compte des variations linguistiques, culturelles et de format. |
| Gestion efficace des changements d'étiquetage | Optimisation des processus de suivi, de mise en œuvre et de documentation des changements d'étiquetage, rapidement et avec précision. |
L'étiquetage multilingue représente un défi majeur pour les entreprises pharmaceutiques opérant sur les marchés mondiaux. Traduire avec précision le contenu de l'étiquetage en plusieurs langues tout en respectant les nuances linguistiques et réglementaires régionales exige une attention particulière aux détails et des processus de gestion de la traduction robustes. Assurer la cohérence et la clarté entre les différentes versions linguistiques est essentiel pour communiquer efficacement des informations vitales à diverses populations de patients.
Maintenir la cohérence entre les portefeuilles de produits présente un autre défi courant en matière d'étiquetage réglementaire. Les entreprises pharmaceutiques gèrent souvent plusieurs produits avec des exigences d'étiquetage, des formulations et des indications différentes. Atteindre la cohérence et la conformité entre diverses gammes de produits tout en répondant aux exigences réglementaires spécifiques de chaque produit nécessite des processus et des systèmes efficaces pour assurer l'uniformité du contenu, du format et des messages de l'étiquetage.
Quelles sont les réglementations clés régissant l'étiquetage pharmaceutique ?
L'étiquetage pharmaceutique est régi par un ensemble complexe de réglementations conçues pour garantir la sécurité, l'efficacité et l'utilisation précise des médicaments. Quelques-unes d'entre elles sont énumérées ci-dessous :
FDA Agence US des produits alimentaires et médicamenteux)
FDA US FDA l'étiquetage des médicaments au moyen d'un ensemble de règles strictes énoncées dans le titre 21 du Code of Federal Regulations (CFR). Ces règles exigent que les étiquettes fournissent des informations complètes, notamment les indications du médicament, les instructions d'utilisation, les contre-indications et les effets secondaires potentiels. La FDA l'importance d'un langage clair, précis et sans ambiguïté afin de garantir la sécurité des patients et de permettre aux professionnels de santé de prendre des décisions en connaissance de cause. En outre, les exigences FDA en matière d'étiquetage s'étendent à divers aspects tels que l'emballage, les notices et l'étiquetage électronique, garantissant ainsi que toutes les informations sont accessibles et normalisées quel que soit le format. Le respect de ces réglementations est obligatoire pour l'autorisation de mise sur le marché et le maintien de la commercialisation d'un médicament aux États-Unis.
EMA (Agence européenne des médicaments)
L'EMA supervise l'étiquetage pharmaceutique dans l'EU (European Union) par le biais de directives et de lignes directrices visant à harmoniser l'étiquetage dans les Member States. La Directive 2001/83/CE de la Commission européenne est essentielle à ces efforts, spécifiant les exigences pour le Résumé des Caractéristiques du Produit (SmPC), les notices d'information pour les patients et les étiquettes d'emballage. L'EMA veille à ce que l'étiquetage inclue des informations essentielles pour les professionnels de la santé et les patients, favorisant une utilisation sûre et efficace des médicaments dans toute l'EU (European Union). De plus, l'étiquetage doit être disponible dans les langues officielles des Member States où le médicament est commercialisé, reflétant l'engagement de l'EMA en faveur de l'accessibilité et des soins centrés sur le patient.
TGA (Therapeutic Goods Administration)
En Australie, la TGA est responsable de la réglementation de l'étiquetage pharmaceutique en vertu de la loi de 1989 sur les produits thérapeutiques (Therapeutic Goods Act 1989). Les directives de la TGA exigent que les étiquettes des médicaments fournissent des informations claires, précises et complètes sur le produit, y compris ses ingrédients, ses indications, sa posologie et ses risques potentiels. Les exigences d'étiquetage sont conçues pour protéger la santé publique en garantissant que les consommateurs et les professionnels de la santé disposent des informations nécessaires pour utiliser les médicaments en toute sécurité et efficacité. La TGA met également un accent particulier sur la lisibilité des étiquettes, exigeant qu'elles soient rédigées en anglais simple et que les informations essentielles soient affichées de manière proéminente pour prévenir les erreurs d'utilisation et de médication.
Santé Canada
Santé Canada réglemente l'étiquetage des produits pharmaceutiques par le biais d'un cadre qui priorise la sécurité et le bien-être des patients et des professionnels de la santé. La Loi sur les aliments et drogues et ses règlements connexes définissent les exigences relatives aux étiquettes des médicaments, qui doivent inclure des informations détaillées sur la composition du produit, les indications, les contre-indications et les effets secondaires potentiels. Santé Canada exige également que les étiquettes soient bilingues, présentées en anglais et en français, afin de tenir compte de la diversité linguistique du pays. De plus, Santé Canada met régulièrement à jour ses exigences en matière d'étiquetage pour refléter les nouvelles preuves scientifiques et les besoins évolutifs en matière de santé publique, garantissant ainsi que l'étiquetage reste pertinent et efficace pour promouvoir une utilisation sûre des médicaments.
PMDA (Agence des produits pharmaceutiques et des dispositifs médicaux)
La PMDA, l'autorité réglementaire japonaise, supervise l'étiquetage pharmaceutique conformément à la Loi sur les affaires pharmaceutiques et aux directives connexes. La PMDA exige que les étiquettes des médicaments fournissent des informations complètes, y compris les indications, les instructions de dosage et les effets indésirables potentiels, dans un format facilement compréhensible par les professionnels de la santé et les patients. La PMDA impose également que les étiquettes incluent des avertissements et des précautions spécifiques à la population japonaise, en tenant compte de facteurs tels que les différences génétiques et les pratiques culturelles. Cette approche garantit que les médicaments sont utilisés en toute sécurité et efficacement au Japon, avec un étiquetage adapté aux besoins uniques du marché local.
NMPA (Administration Nationale des Produits Médicaux)
En Chine, la NMPA régit l'étiquetage pharmaceutique par le biais d'un cadre réglementaire qui met l'accent sur l'exactitude, la clarté et la sécurité. La Loi sur l'administration des médicaments de la République populaire de Chine énonce les exigences relatives aux étiquettes des médicaments, qui doivent inclure des informations sur les indications, la posologie, les contre-indications et les effets secondaires potentiels du médicament. La NMPA exige également que l'étiquetage soit présenté en chinois simplifié afin d'assurer l'accessibilité à la population locale. De plus, la NMPA exige que les étiquettes fassent l'objet d'un examen rigoureux pendant le processus d'approbation des médicaments afin d'assurer la conformité aux normes nationales et de protéger la santé publique en prévenant les erreurs de médication et les abus.
Comment un partenaire réglementaire peut-il aider à se conformer aux exigences d'étiquetage ?
Un partenaire spécialisé en réglementation joue un rôle essentiel pour garantir la conformité aux exigences en matière d'étiquetage, grâce à son expertise pointue et à son accompagnement complet. Il guide les entreprises à travers le paysage réglementaire complexe, en veillant à ce que les supports d'étiquetage — notamment les emballages, les notices et les étiquettes électroniques — respectent les exigences spécifiques des différentes autorités sanitaires telles que laFDA US , EMA, la TGA, Santé Canada, PMDA et NMPA. Cela implique de comprendre et d'appliquer les dernières réglementations, qui peuvent varier considérablement d'une région à l'autre, afin de garantir que toutes les informations sur les produits sont exactes, complètes et conformes.
De plus, un partenaire réglementaire aide à simplifier le processus d'étiquetage en fournissant des services essentiels tels que la création, la révision et la validation de contenu. Il aide à rédiger et à réviser le contenu de l'étiquetage pour l'aligner sur les normes réglementaires et s'assure que toutes les informations nécessaires sont incluses, des listes d'ingrédients et instructions d'utilisation aux avertissements de sécurité et conditions de stockage. Cela réduit le risque d'erreurs et d'omissions qui pourraient entraîner des retards réglementaires ou des retraits du marché, accélérant ainsi la mise sur le marché de nouveaux produits.

De plus, un partenaire réglementaire aide les entreprises à maintenir une conformité continue en surveillant les mises à jour réglementaires et en mettant en œuvre les changements nécessaires. Il offre des conseils stratégiques pour adapter les étiquettes aux nouvelles directives ou aux exigences des marchés émergents, aidant ainsi les entreprises à éviter les problèmes de non-conformité et à garantir que leurs produits restent conformes aux réglementations en vigueur. En tirant parti de son expertise et en se tenant au courant des changements réglementaires, un partenaire réglementaire aide les entreprises à naviguer efficacement dans le paysage dynamique de l'étiquetage.
Comment les entreprises peuvent-elles démarrer avec les services d'étiquetage réglementaire ?
Pour démarrer avec les services d'étiquetage réglementaire, les entreprises doivent d'abord évaluer leurs besoins spécifiques en matière d'étiquetage en fonction des marchés cibles et des exigences réglementaires. Ensuite, elles doivent s'associer à un fournisseur de services réglementaires fiable, expert en normes d'étiquetage mondiales. Ce fournisseur peut aider à la rédaction, à l'examen et à la mise à jour des étiquettes pour garantir la conformité. De plus, la mise en œuvre d'un système centralisé de Management du labelling permet de rationaliser le processus, assurant la cohérence entre toutes les étiquettes de produits. Des audits et des mises à jour réguliers sont essentiels pour maintenir les étiquettes en conformité avec les réglementations en évolution.
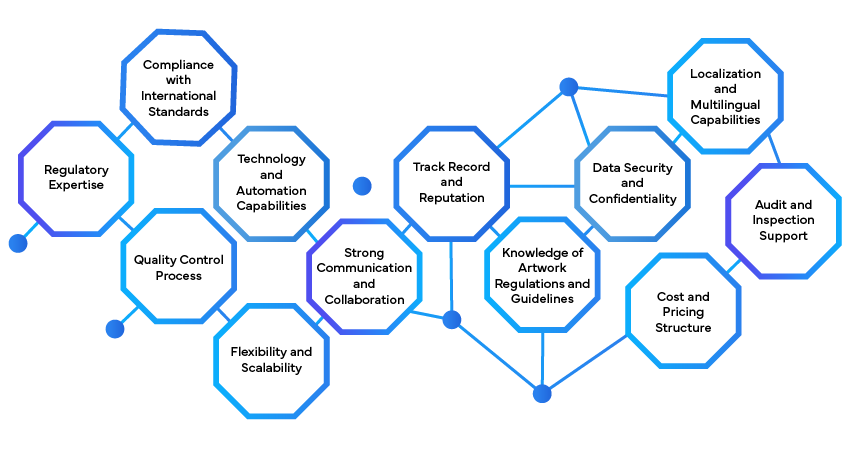
Les services d'étiquetage réglementaire peuvent-ils aider à la surveillance post-commercialisation ?
Oui, les services d'étiquetage réglementaire peuvent effectivement aider au suivi après commercialisation des produits pharmaceutiques. Ces services jouent un rôle crucial en soutenant la surveillance après commercialisation, en facilitant la gestion des mises à jour d'étiquetage, en traitant les modifications d'étiquetage liées à la sécurité et en garantissant la conformité aux exigences réglementaires post-approbation. En maintenant des informations d'étiquetage précises et à jour, les services d'étiquetage réglementaire aident les entreprises pharmaceutiques à répondre aux préoccupations de sécurité et à mettre en œuvre les changements nécessaires en temps voulu pour soutenir la conformité réglementaire continue et la sécurité des patients.
De plus, les services d'étiquetage réglementaire peuvent faciliter la diffusion efficace des informations de sécurité mises à jour aux professionnels de la santé et aux patients. En cas de nouvelles découvertes en matière de sécurité ou de modifications des profils de risque pour les produits pharmaceutiques, les experts en étiquetage réglementaire peuvent aider à mettre à jour rapidement le contenu de l'étiquetage pour refléter les dernières données de sécurité et les exigences réglementaires. Cette approche proactive de la surveillance après commercialisation et des mises à jour de l'étiquetage soutient la communication rapide d'informations de sécurité importantes aux prestataires de soins de santé et aux patients, contribuant ainsi à une pharmacovigilance et à des soins aux patients améliorés.
Dans l'ensemble, les services de Management du labelling réglementaire offrent un soutien précieux pour la surveillance après commercialisation en veillant à ce que les produits pharmaceutiques maintiennent un labelling précis et conforme tout au long de leur cycle de vie. En tirant parti de l'expertise des professionnels de la réglementation et de processus efficaces de gestion du labelling, les entreprises peuvent gérer efficacement les considérations de sécurité post-commercialisation et les obligations réglementaires, contribuant ainsi à la sécurité et à l'efficacité continues de leurs produits sur le marché.
Pourquoi choisir Freyr ?
![about-us]()
Une décennie d'excellence en matière d'étiquetage réglementaire.
![about-us]()
Plus de 180 experts mondiaux en services d'étiquetage
![about-us]()
Se spécialise dans la création et la gestion de documents essentiels
![about-us]()
Expertise en matière de brochures pour investigateurs, de fiches de données essentielles et de fiches de données essentielles de l'entreprise
![about-us]()
Engagés envers la conformité mondiale et la précision
![about-us]()
Utilise l'IA (Intelligence Artificielle) pour une navigation réglementaire efficace






Collaborez avec nous dès maintenant
En bref








Foire aux questions
Les fiches de données essentielles (CDS) offrent un résumé consolidé des informations critiques sur les médicaments, incluant les indications, les posologies et les profils de sécurité. Elles assurent une communication cohérente des détails essentiels sur les marchés mondiaux, facilitant la conformité réglementaire et la prise de décision éclairée. Les CDS servent également de référence pour la création des étiquettes de produits locaux.
Les brochures de l'investigateur (IB) détaillent les données des essais cliniques et les informations sur le développement de médicaments à usage expérimental, tandis que les Company Core Data Sheets (CCDS) résument les données clés de sécurité et d'efficacité à des fins réglementaires mondiales, guidant le contenu et les mises à jour des étiquettes. Les CCDS sont utilisées pour créer des étiquettes spécifiques aux produits en vue de leur approbation sur le marché.
L'intelligence artificielle améliore l'étiquetage réglementaire en automatisant l'analyse des données, en améliorant la précision de la création de contenu et en accélérant l'examen des documents. Les outils d'IA simplifient les processus d'étiquetage et garantissent la cohérence des diverses exigences réglementaires. Ils aident également à prévoir et à résoudre les problèmes potentiels de conformité.
L'étiquetage multilingue garantit que les produits pharmaceutiques sont accessibles à diverses populations de patients, en répondant aux exigences réglementaires régionales et en améliorant la sécurité grâce à des instructions et des avertissements clairs et compréhensibles en plusieurs langues. Cela réduit le risque de mauvaise communication et d'erreurs dans l'administration des médicaments.
Un système centralisé de Management du labelling coordonne la création, la révision et la mise à jour des documents de labelling, assurant cohérence et conformité sur les marchés mondiaux. Il simplifie les processus et maintient des informations produit précises et à jour. Ce système prend également en charge la gestion efficace des modifications de labelling et des mises à jour réglementaires.
Le Structured Product Labeling (SPL) est un format basé sur le XML utilisé pour l'étiquetage des médicaments qui standardise et organise les informations sur les produits. Il assure la cohérence et facilite l'échange de données entre les agences de réglementation et les fabricants. Le SPL prend en charge la gestion efficace des informations d'étiquetage tout au long du cycle de vie d'un produit.
Le Numéro de Localisation Mondial (GLN) est un identifiant unique utilisé pour identifier les lieux et les entités au sein de la chaîne d'approvisionnement. Il aide à suivre et à gérer avec précision les produits pharmaceutiques sur les marchés mondiaux. Les GLN garantissent une distribution des produits et une gestion des stocks précises et efficaces.
Le Code national des médicaments (NDC) est un identifiant unique attribué aux médicaments par la FDA. Il permet d'identifier avec précision les produits pharmaceutiques et facilite la gestion des stocks ainsi que le suivi. Le NDC est essentiel pour garantir l'exactitude de la délivrance des médicaments et pour l'établissement des rapports réglementaires.
Une Brochure pour l'investigateur (IB) fournit des informations détaillées sur les données cliniques et précliniques d'un médicament expérimental. Elle est utilisée pour informer les investigateurs d'essais cliniques sur la sécurité, l'efficacité et la posologie du médicament à des fins d'étude. L'IB soutient également la prise de décision éthique et éclairée dans la recherche clinique.