
Vue d'ensemble de l'approbation pré-commercialisation des dispositifs médicaux par l'USFDA
La procédure d'autorisation USFDA (PMA) USFDA FDA US est l'une des voies d'enregistrement des dispositifs proposées par laFDA, principalement destinée aux dispositifs médicaux FDA III. La procédure d'autorisation FDA pour les dispositifs de classe III implique des évaluations scientifiques et réglementaires minutieuses visant à évaluer la sécurité et l'efficacité du dispositif médical, afin de garantir le respect des normes les plus strictes avant l'autorisation de mise sur le marché.
Réservez une réunion avec nos experts en autorisation de mise sur le marché
Qui devrait soumettre une demande d'approbation préalable à la mise sur le marché (PMA) pour dispositif médical auprès de l'USFDA ?
Les fabricants de dispositifs doivent soumettre une demande de PMA si le dispositif :
- Est innovant.
- Appartient à une classe à risque élevé.
- Introuvable dans la base de données de classification des produits.
- N'est pas substantiellement équivalent (NSE) aux dispositifs de Classe I, II ou III.
Obtenez des conseils d'experts sur votre demande d'autorisation de mise sur le marché
Quelle est la différence entre les demandes 510(k), PMA et De-Novo ?
Approbation précommercialisation
- Dispositif de classe III qui soutient la vie humaine ou qui présente un risque potentiel et déraisonnable de maladie ou de blessure.
- La procédure d'autorisation FDA nécessite la réalisation d'essais cliniques.
- Nécessite une inspection sur site avant la délivrance de l'approbation PMA.
- 180 jours calendaires
Classification De Novo
- Nouveaux dispositifs de Classe I et II qui n'ont pas de dispositif de référence valide.
- Nécessite des données d'études cliniques.
- Aucun audit sur site avant l'approbation De-Novo.
- 150 jours calendaires.
Enregistrement 510(k)
- Dispositifs FDA III FDA présentant une équivalence substantielle avec le dispositif de référence.
- Cela ne nécessite pas d'essais sur l'homme.
- Aucun audit sur site avant l'autorisation 510(k).
- 90 jours calendaires.
Quelles sont les différentes méthodes de demande d'autorisation FDA ?
Les fabricants peuvent opter pour l'une des quatre (04) méthodes de demande PMA suivantes, la mieux adaptée à leur dispositif :
- PMA traditionnelle
- PMA modulaire
- Protocole de développement de produits
- Exemption pour dispositif humanitaire
Quelles sont les exigences en matière de données pour l'approbation préalable à la mise sur le marché des dispositifs médicaux ?
Conformément à la partie 814 du titre 21 du Code of Federal Regulations (CFR), les demandeurs doivent soumettre à laFDA US un formulaire de demande du CDRH dûment rempli, les engagements requis et un dossier technique PMA bien rédigé. Le dossier technique doit inclure les données non cliniques et cliniques.
Données non cliniques – Elles consistent en des données sur la microbiologie, la toxicologie, l'immunologie, la biocompatibilité, le stress, l'usure, la durée de conservation et d'autres tests de laboratoire ou sur animaux.
Données cliniques – Elles comprennent des données sur les protocoles d'étude, les données de sécurité et d'efficacité, les réactions indésirables et les complications, les défaillances et remplacements de dispositifs, les informations sur les patients, les plaintes des patients, les tableaux de données de tous les sujets individuels, les résultats des analyses statistiques et toute autre information provenant des investigations cliniques.
Qu'est-ce que le processus de demande PMA ?
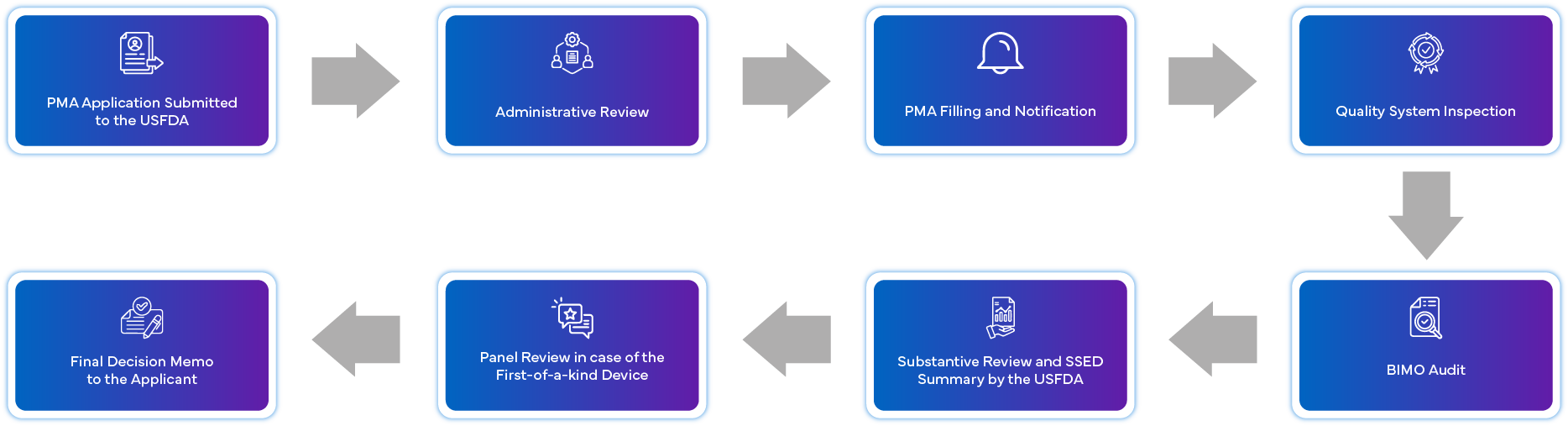
Quelles sont les exigences de conformité après approbation pour le PMA ?
Les dispositifs approuvés via la voie PMA doivent se conformer aux exigences post-commercialisation établies par l'USFDA. Le dispositif doit se conformer aux éléments suivants :
- Exigences post-autorisation imposées dans la décision d'autorisation de mise sur le marché ( FDA .
- Gestion des changements post-approbation grâce à la soumission rapide des suppléments PMA pertinents
- Soumission des rapports post-approbation (annuels)
- Réglementations 21 CFR 803 pour la déclaration des dispositifs médicaux (MDR)
- Études de surveillance post-commercialisation telles qu'exigées par la USFDA dans les ordres d'approbation PMA.
Quels sont les frais de l'USFDA pour l'examen de la demande PMA ?
Les frais d'utilisation MDUFA pour les PMA originaux et les suppléments sont les suivants –
| Type de demande | Frais pour l'exercice fiscal 2023 (du 1er octobre 2022 au 30 septembre 2023) | |
|---|---|---|
| Frais standard | Frais pour les petites entreprises | |
| PMA, PDP, PMR, BLA | $441,547 |
|
| Supplément de voie de panel | $353,238 | $88,309 |
| Supplément de 180 jours | $66,232 | $16,558 |
| Frais annuels pour les rapports périodiques sur un dispositif de Classe III (PMA, PDP et PMR) | $15,454 | $3,864 |
| Préavis de 30 jours | $7,065 | $3,532 |
| Supplément en temps réel | $30,908 | $7,727 |
Grâce à son expertise dans la gestion des soumissions PMA, Freyr peut aider à identifier et à compiler les informations, ainsi qu'à la préparation et à l'examen de la demande.
Expertise et avantages de l'USFDA en matière d'approbation pré-commercialisation des dispositifs médicaux
- Diligence raisonnable réglementaire
- Conformité aux Inspections du Système Qualité
- Conformité aux audits BIMO
- Compilation du dossier technique PMA
- Publication et création d'eCopy
- Validation et soumission de l'eCopy
- Traite les réponses aux RTA et les lacunes.
- Services d'accompagnement jusqu'à l'autorisation FDA
- Consultation pour les déficiences
- Enregistrement des dispositifs et des établissements
- Gestion des suppléments PMA et des notifications de 30 jours
- Soumissions de rapports périodiques annuels
- Audits blancs et formation 21 CFR 820

- Expérience dans le traitement de nombreuses demandes FDA pour diverses catégories de dispositifs médicaux.
- Équipe d'experts spécialisée dans les demandes d'autorisation FDA , conformément aux exigences réglementaires
- Soutien supplémentaire pour gérer les demandes liées aux PMA.
- Soumission dans les délais des livrables
- À jour avec lesFDA modifications apportées par laFDA US
