3 minutes de lecture
BIMO signifie « Bioresearch Monitoring » ; il s'agit d'un programme d'inspections sur site et d'audits de données visant à contrôler tous les aspects de la conduite et de la communication des résultats des recherches réglementées par US and Drug Administration (FDA) US . Ce programme a été mis en place en 1977 après qu'un besoin d'auditer les sites de recherche clinique a été identifié. L'objectif principal de ce programme est de garantir la qualité et l'intégrité des données soumises dans le cadre des demandes d'autorisation de mise sur le marché et d'homologation de nouveaux produits. En outre, ce programme protège également les droits et le bien-être des sujets humains et animaux participant à la recherche FDA.
Les objectifs clés du programme BIMO
Chaque année, plus de 1000 inspections sont menées. Les principaux objectifs couverts par le programme BIMO sont :
- Auditer les données cliniques
- Inspection de la recherche clinique en cours
- Inspection des laboratoires non cliniques
- Inspection des Comités d'éthique de la recherche (IRB)
Quels produits relèvent du champ d'application de l'audit BIMO ?
Le BIMO s'applique aux médicaments, aux produits biologiques, aux dispositifs médicaux, aux produits alimentaires, aux produits du tabac et aux produits vétérinaires. Le programme de conformité est supervisé par les six (06) centres de produits FDA: le Centre d'évaluation et de recherche sur les produits biologiques (CBER), le Centre des dispositifs médicaux et de la santé radiologique (CDRH), le Centre d'évaluation et de recherche sur les médicaments (CDER), le Centre de la sécurité alimentaire et de la nutrition appliquée (CFSAN), le Centre des produits du tabac (CTP) et le Centre de médecine vétérinaire (CVM).
Quelles entreprises sont soumises à un audit BIMO ?
Les entreprises nationales et internationales qui exercent ou relèvent de l'une des activités ci-dessous sont soumises aux exigences de la Surveillance de la Biorécherche -
- Laboratoires d'essais non cliniques pour la conformité aux Bonnes Pratiques de Laboratoire (GLP)
- Investigateurs cliniques pour la conformité aux Bonnes Pratiques Cliniques (GCP)
- Promoteurs
- Organisations de Recherche Contractuelle (CRO)
- Attachés de recherche clinique (ARC)
- Installations de bioéquivalence in vivo
- Comités d'éthique de la recherche (IRB)
Quels programmes de conformité relèvent du programme BIMO ?
FDA US FDA à tout moment mener un audit BIMO dans le cadre des sept (07) programmes de conformité multicentriques. Ces sept programmes de conformité multicentriques sont mis en œuvre par le biais de –
- Inspection des investigateurs cliniques (IC) et des investigateurs-promoteurs (IP)
- Inspection des Comités d'éthique de la recherche (IRB)
- Inspection d'une Organisation de Recherche Contractuelle/Promoteur/Moniteur (CRO/P/M)
- Inspection des Bonnes Pratiques de Laboratoire (GLP)
- Inspection de bioéquivalence-biodisponibilité (BEQ)
- Inspection des rapports d'expériences indésirables médicamenteuses après commercialisation (PADE)
- Inspection des rapports d'évaluation des risques, mesures d'atténuation et stratégie (REMS)
Chacun de ces programmes définit le champ d'application détaillé de l'examen ou de l'inspection à mener afin de garantir la conformité aux exigences de FDA.
Quelles réglementations sont applicables pour l'audit BIMO ?
Les réglementations – 21 CFR 50 – Protection des sujets humains, 21 CFR 54 – Divulgation financière, 21 CFR 56 – IRBs, 21 CFR 58 – Bonnes pratiques de laboratoire pour les laboratoires non cliniques, 21 CFR 809 – Produits de diagnostic in vitro et 21 CFR 812 – Exemption pour les dispositifs expérimentaux sont applicables pour l'audit BIMO.
Combien d'audits sont réalisés chaque année dans le cadre du programme BIMO ?
Le nombre d'audits BIMO menés par laFDA US FDA d'une année à l'autre. Ces dernières années, le nombre d'inspections sur site a diminué en raison de la pandémie de COVID-19, et la FDA dû suspendre toute surveillance sur site des essais cliniques. Seuls certains essais cliniques jugés critiques et essentiels faisaient l'objet d'un suivi.
Les « Évaluations Réglementaires à Distance » (RRAs) ont été introduites pendant la pandémie de COVID-19 pour surveiller à distance la recherche réglementée. Les RRAs sont menées par vidéoconférence et constituent une initiative volontaire pour évaluer à distance les données et les processus. Cependant, il convient de noter que les RRAs ne sont pas équivalentes ou une alternative à l'inspection sur site, mais sont simplement une procédure qui a évolué en raison de la pandémie de COVID-19.
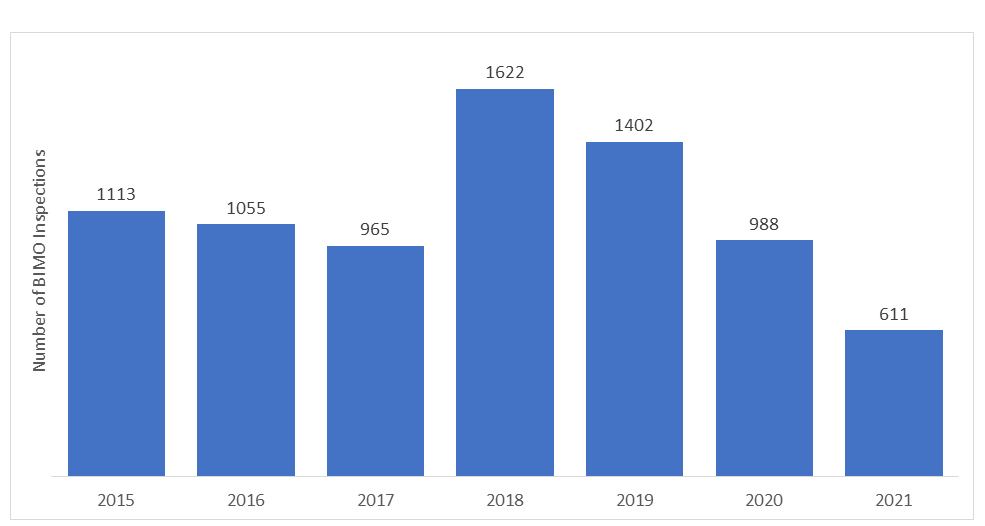
*Les données représentées pour les années 2020 et 2021 n'incluent pas les inspections RRA
Combien d'évaluations réglementaires à distance (RRA) ont été menées pendant la pandémie de COVID-19 dans le cadre du programme BIMO ?
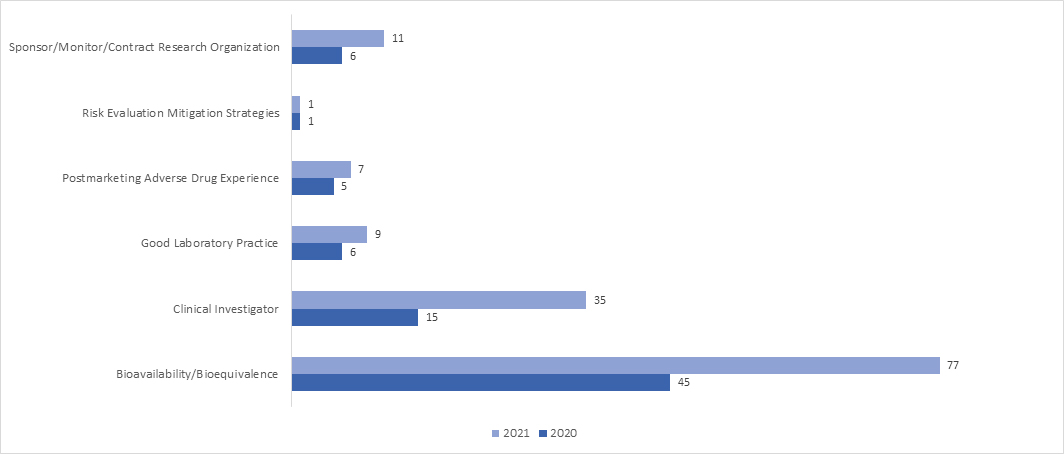
En 2021, le recours aux inspections RRA a connu une forte augmentation dans l’ensemble des programmes. En avril 2021, la FDA un document d’orientation intitulé « Évaluations interactives à distance des installations de fabrication de médicaments et de surveillance de la recherche biologique pendant l’urgence de santé publique liée à la COVID-19 : lignes directrices à l’intention de l’industrie », qui fournit des informations exhaustives sur la procédure FDApour mener ces inspections RRA.
Quels sont les résultats possibles d'un audit BIMO ?
Au cours de l'audit BIMO, laFDA US FDA décider de prendre l'une des mesures énumérées ci-dessous en fonction du niveau de conformité –
1. Aucune action indiquée (NAI)
La NAI s'applique lorsque l'inspecteur FDA n'a constaté aucune pratique répréhensible ou n'a relevé que des problèmes mineurs ne justifiant pas la prise de mesures supplémentaires.
2. Action volontaire indiquée (VAI)
Le VAI est applicable lorsque des pratiques répréhensibles ont été identifiées mais ne sont pas significatives.
3. Action officielle indiquée (OAI)
L'OAI est applicable lorsque des pratiques répréhensibles qui compromettent l'intégrité des données et/ou les droits des sujets humains sont identifiées.
Quelles sont les non-conformités les plus courantes émises dans le cadre d'un audit BIMO ?
Certaines des non-conformités les plus courantes observées lors de l'audit BIMO sont :
- Ne pas tenir un suivi approprié des enregistrements
- Manquement concernant le plan d'investigation
- Non-conformité aux réglementations
- Manquement dans le suivi des protocoles
- Protection insuffisante des sujets
- Manque de responsabilité concernant le produit faisant l'objet de l'enquête
L'audit BIMO est crucial pour tout développeur ou fabricant de dispositifs médicaux et technologies innovants qui prévoit de lancer son dispositif sur le marché US. Il est très important de se conformer aux réglementations et aux directives pour éviter les pièges décrits.
Avez-vous besoin d'aide concernant les inspections d'audit BIMO ? Contactez Freyr. Restez informé. Restez conforme.