

3 minutes de lecture
Entreprendre un essai clinique est une étape majeure dans le développement d'un produit médicamenteux ou d'un dispositif médical. Une demande d'autorisation d'essai clinique (CTA) est nécessaire pour que les entreprises pharmaceutiques ou les promoteurs obtiennent l'approbation des autorités réglementaires afin de commencer des études chez l'homme. À mesure que la recherche clinique devient plus mondiale et complexe, il est crucial de comprendre les pratiques internationales en évolution, les étapes clés de la soumission et les avantages offerts par un ou plusieurs partenaires en affaires réglementaires (RA) pour les promoteurs comme pour les organismes de recherche.
Demande d'autorisation d'essai clinique (CTA) et ses sections :
Une demande d'autorisation d'essai clinique (CTA) est le dossier réglementaire que les promoteurs doivent soumettre aux autorités réglementaires nationales (ARNs) avant de débuter une étude clinique impliquant des participants humains. Les CTA sont obligatoires pour la plupart des essais interventionnels de médicaments, de dispositifs médicaux ou de produits biologiques. Elles garantissent que l'essai est scientifiquement valide, éthiquement approprié et conforme aux directives locales et internationales, telles que les normes de Bonnes Pratiques Cliniques (GCP) de l'International Council for Harmonization (ICH) et les exigences nationales.
Les principales sections d'une CTA typique comprennent (liste non exhaustive) :
- Lettre d'accompagnement et formulaires de demande
- Protocole d'étude clinique et brochure de l'investigateur
- Dossier du produit et preuves de Bonnes Pratiques de Fabrication (BPF)
- Approbations éthiques et du Comité d'examen institutionnel (IRB)
- Assurance et documentation sur la protection des participants
- Plan de surveillance de la sécurité des données et déclarations financières
Pratiques mondiales pour les soumissions de CTA
Les processus et les exigences des CTA varient selon les régions, mais plusieurs principes communs se dégagent :
1. Harmonisation des formats et des normes
- De nombreux pays utilisent le document technique commun (CTD) de l'ICH, ce qui simplifie les soumissions d'études multinationales. Sa structure modulaire facilite l'examen parallèle et simplifie les mises à jour.
- Des adaptations locales sont courantes, notamment dans les régions Asie-Pacifique et Pacifique occidental, où les pays combinent souvent le CTD avec des formulaires nationaux, des retours d'examen, des besoins linguistiques ou des études passerelles.
2. Consultation réglementaire
- Un engagement précoce avec les agences — telles que la FDA US, les autorités nationales de l'UE et les agences au Japon, en Chine et sur les principaux marchés émergents — est encouragé, en particulier pour les essais cliniques mondiaux et multirégionaux (MRCTs). Les réunions de conseils scientifiques réduisent les rejets futurs et orientent l'optimisation des protocoles.
3. Voies collaboratives et accélérées
- Les agences acceptent de plus en plus la « reliance » ou les évaluations collaboratives, où elles s'appuient sur les examens ou les approbations d'autorités réglementaires strictes, ce qui accélère les processus et assure une meilleure cohérence.
- Des options d'examen accéléré ou de procédure d'urgence peuvent être disponibles, en particulier lors d'urgences sanitaires ou pour les thérapies innovantes.
4. Intégration de l'examen éthique
- Dans l'UE et de nombreuses autres régions, les examens éthiques et réglementaires peuvent avoir lieu en parallèle ou via des plateformes coordonnées afin d'éviter des soumissions séparées et de réduire les délais de démarrage.
5. Transparence et publication
- Il est désormais courant d'enregistrer les essais dans des bases de données reconnues avant leur approbation, et de nombreux pays publient l'état d'approbation des CTA, contribuant ainsi à la transparence mondiale et aux meilleures pratiques.
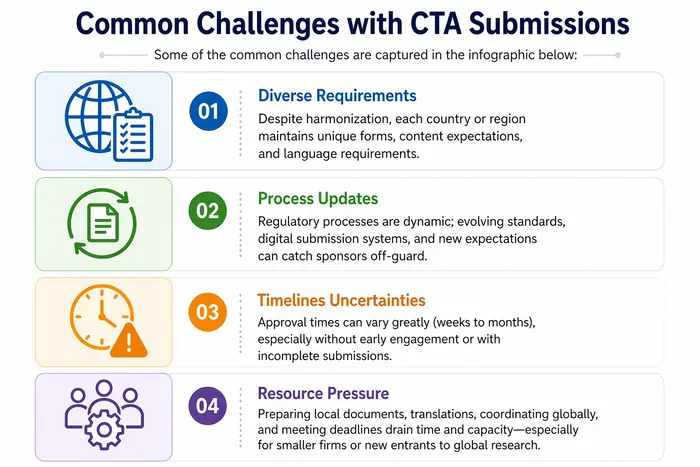
Comment un partenaire comme Freyr peut-il vous aider ?
Un partenaire de confiance en affaires réglementaires (RA) comme Freyr offre un soutien continu et une atténuation des risques tout au long du processus de CTA. Voici pourquoi l'engagement d'un tel expert assure le succès des promoteurs :
1. Veille réglementaire mondiale actualisée
- Nous surveillons en permanence les changements réglementaires, les interprétations régionales et les pratiques de soumission dans le monde entier. Cela garantit que chaque CTA est élaborée selon les exigences les plus récentes, minimisant ainsi les questions, les rejets et les retards coûteux des essais.
2. Préparation et examen du dossier
- Les spécialistes de Freyr élaborent et compilent les CTA en utilisant les derniers formats ICH CTD et des modèles nationaux adaptés. Ils coordonnent les traductions de documents, organisent les réponses aux dossiers et garantissent une qualité uniforme et la conformité aux GCP pour chaque soumission.
3. Consultation réglementaire et liaison avec les agences
- Freyr peut représenter les promoteurs lors des réunions de conseil scientifique pré-CTA, gérer les questions des agences après la soumission et soutenir les communications avec les autorités et les comités d'éthique.
4. Gestion de projet mondiale
- Avec des équipes multiculturelles expérimentées et des outils de gestion de projet robustes, Freyr coordonne les soumissions simultanées, suit les progrès pays par pays et assure l'harmonisation pour les MRCT (essais cliniques multirégionaux).
5. Stratégie de reliance et d'examen accéléré
- Freyr aide les promoteurs à tirer parti des cadres de reliance, à préparer des dossiers abrégés pour des examens dispensés ou accélérés, et à harmoniser les données de soumission pour une acceptation réglementaire maximale.
6. Préparation aux audits et au contrôle des changements
- Le partenaire s'assure que la documentation et les procédures des promoteurs respectent les normes d'audit, facilitant ainsi une approbation réglementaire rapide et des inspections sans heurts.
Conclusion
La soumission d'une CTA est une étape complexe mais essentielle pour la recherche clinique, avec des exigences mondiales et spécifiques à chaque région. Les tendances mondiales incluent l'harmonisation du CTD, l'examen éthique et réglementaire parallèle, les mécanismes de reliance et les nouvelles normes de transparence. Les promoteurs sont confrontés à des défis notables pour répondre à des exigences diverses, suivre le rythme des changements réglementaires et gérer les délais mondiaux. Dans un tel scénario, un partenaire en affaires réglementaires tel que Freyr offre une expertise End-to-End en veille réglementaire, préparation de dossiers, interactions avec les agences, voies accélérées et préparation aux audits, augmentant considérablement les chances de démarrer des essais cliniques avec succès et dans les délais.









