Compliance bei der Registrierung von Medizinprodukten und die regulatorischen Rahmenbedingungen in der EU
Die Europäische Union stellt einen der am stärksten regulierten und wirtschaftlich attraktivsten Märkte für Medizinprodukte weltweit dar. Mit 27 member states, 3 EEA und der Türkei unterliegt der EU-Markt einem harmonisierten Regulierungsrahmen, der durch EU MDR 2017/745) für Medizinprodukte und die EU-IVDR (2017/746) für In-vitro-Diagnostika definiert ist; damit gewährleistet die Region einheitliche Sicherheits- und Leistungsstandards für alle Produktkategorien. Um ein Produkt rechtmäßig auf dem EU-Markt in Verkehr zu bringen, müssen Hersteller die CE-Kennzeichnung erhalten, die die Konformität mit den geltenden Vorschriften auf der Grundlage der Risikoklassifizierung des Produkts und des Konformitätsbewertungsverfahrens bestätigt. Die Nichteinhaltung dieser Anforderungen kann schwerwiegende Folgen haben, darunter Produktrückrufe, Beschlagnahmung am Zoll, Aussetzung der Zertifizierung oder der vollständige Verlust des Marktzugangs.
Nach dem derzeitigen Regulierungssystem:
- Die CE-Kennzeichnung ist für alle Medizinprodukte und In-vitro-Diagnostika vorgeschrieben, bevor sie in Verkehr gebracht werden dürfen.
- Die EUDAMED-Produktregistrierung wird schrittweise eingeführt, wobei mehrere Module bereits verpflichtend genutzt werden müssen.
- Die Benennung eines europäischen Bevollmächtigten (EAR) ist für jeden Hersteller mit Sitz außerhalb der EU, desEEA verpflichtend.
- Strengere Klassifizierungsvorschriften gemäß MDR/IVDR erfordern eine fundierte technische Dokumentation, klinische oder leistungsbezogene Nachweise sowie eine kontinuierliche Überwachung während des gesamten Lebenszyklus.
Freyr unterstützt Hersteller auf diesem Weg, indem es ihnen einen reibungslosen und vorschriftsmäßigen Zugang zum EU-Markt ermöglicht. Von der Entwicklung einer Strategie für die CE-Kennzeichnung über die Erstellung der technischen Dokumentation bis hin zur Abwicklung der Anträge bei benannten Stellen und der Übernahme der Rolle Ihres EU-Bevollmächtigten sorgt Freyr für eine lückenlose Einhaltung der MDR/IVDR-Vorschriften und einen nahtlosen Zugang zu allen member states,EEA und der Türkei.
Schritt-für-Schritt-Anleitung zur Einhaltung der EU-Vorschriften
Die Marktzulassung eines Medizinprodukts oder In-vitro-Diagnostikums auf dem EU-Markt umfasst mehrere festgelegte Phasen. So wickelt Freyr den gesamten Prozess für Sie ab:
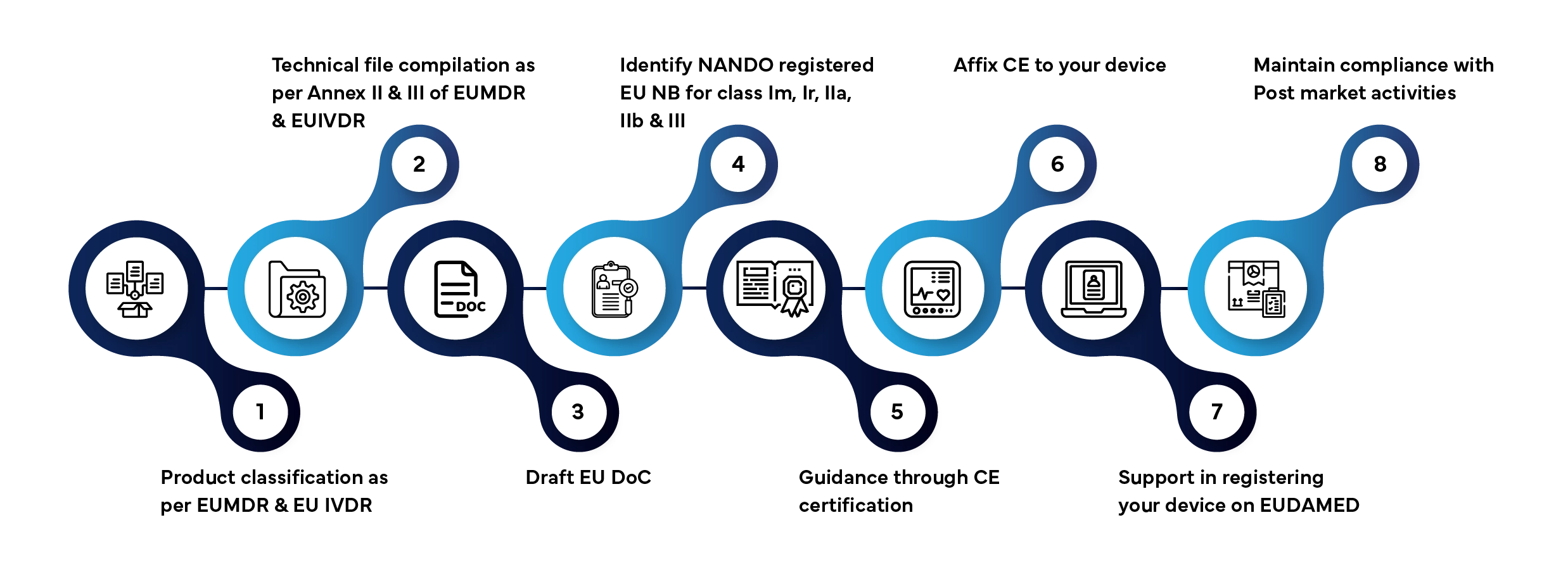
Übliche Bearbeitungszeit: 3–12 Monate, abhängig von der Produktklasse und der Vollständigkeit der Unterlagen.
Die wichtigsten Angebote von Freyr Medical Device in der EU
- Regulierungsstrategie und Übergang zu MDR/IVDR – Wir erstellen maßgeschneiderte, end-to-end für die EU MDR die EU-IVDR und sorgen so für einen reibungslosen Übergang von den bisherigen Richtlinienregelungen sowie für die Anpassung an die aktuellen EU-Anforderungen.
- Technische Dokumentation und Konformitätsbewertung – Unser Team unterstützt Sie bei der Erstellung, Überprüfung und Einreichung Ihrer technischen Unterlagen bzw. Konstruktionsdossiers, begleitet Sie bei der Zusammenarbeit mit benannten Stellen und kümmert sich um die CE-Kennzeichnung sowie um die Unterstützung bei Sicherheits- und Leistungsprüfungen von Medizinprodukten und die Konformitätsverfahren.
- Klinische Bewertung/Leistungsbewertung: Freyr bietet fachkundige Erstellung von CERs, PERs, PMCF, PSURs, SVRs, CPRs für In-vitro-Diagnostika, Risikodokumentationen sowie Inhalten zur biologischen Bewertung und gewährleistet dabei technische Klarheit und regulatorische Genauigkeit für alle Produktklassen.
- Unterstützung bei der UDI- und EUDAMED-Registrierung – Wir stellen sicher, dass Ihr System zur eindeutigen Produktkennzeichnung (UDI) den Vorschriften entspricht, und unterstützen Sie bei der Registrierung in der europäischen Medizinproduktedatenbank EUDAMED sowie beim damit verbundenen Lebenszyklusmanagement.
- Beauftragter in der EU (EAR) & Vertretung vor Ort – Für Hersteller außerhalb derEEA fungieren wir als Ihr beauftragter Vertreter in der EU (EAR) und bieten Ihnen Unterstützung bei der Einhaltung der Vorschriften in allen member states.
- Marktüberwachung (PMS) – Freyr unterstützt Sie bei der Einrichtung, Umsetzung und Pflege von PMS-Systemen, einschließlich des Marktüberwachungsplans (PMSP), Berichte zur Marktüberwachung (PMSR), regelmäßige Sicherheitsberichte (PSUR), Pläne zur klinischen Nachbeobachtung und Leistungsüberwachung nach dem Inverkehrbringen (PMCF), Vigilanzberichte, FSCAs, FSNs sowie CAPAs, um die kontinuierliche Aufrechterhaltung der CE-Kennzeichnung und einen nachhaltigen Marktzugang unter Einhaltung der Anforderungen während des gesamten Produktlebenszyklus sicherzustellen.
- CE-Registrierung von Medizinprodukten: Freyr begleitet den gesamten Prozess der CE-Kennzeichnung in der EU, indem das Unternehmen die Konformitätsbewertung unterstützt, konforme Anträge zusammenstellt, mit benannten Stellen zusammenarbeitet und für eine zeitnahe Zulassung in allen Kategorien von Medizinprodukten und In-vitro-Diagnostika sorgt.
- QMS-Unterstützung: Wir unterstützen die Einführung und Aufrechterhaltung von Qualitätsmanagementsystemen ISO 13485, die den Qualitäts- und Sicherheitsanforderungen EU MDR sowie den Erwartungen der benannten Stellen entsprechen.
- Einhaltung der Kennzeichnungsvorschriften: Unser Team stellt sicher, dass Ihre Kennzeichnungen, Gebrauchsanweisungen, Verpackungen und Symbole den Anforderungen der MDR/IVDR sowie den mehrsprachigen Vorschriften der EU entsprechen, und gewährleistet so Einheitlichkeit und Konformität in allen 27 member states.
Dienstleistungsangebot für EU-Bevollmächtigte (EAR)

Registrierung von Medizinprodukten bei den EU-Behörden
Für Hersteller außerhalb der EU fungiert Freyr als benannter Bevollmächtigter in der EU (EAR) gemäß den Anforderungen der MDR/IVDR. Als in Deutschland ansässiges Unternehmen unterstützt Freyr die Registrierung von Medizinprodukten bei der zuständigen deutschen Behörde und führt die erforderlichen Aufzeichnungen. Für andere Member States bietet Freyr gegebenenfalls regulatorische Unterstützung an, um die Konformität der Medizinprodukte sicherzustellen und deren Inverkehrbringen auf dem EU-Markt zu erleichtern.

Dokumentation und Konformitätssicherung
Unsere Experten für Rechtsvorschriften prüfen, ob Ihre Konformitätserklärung (DoC), Ihre CE-Zertifikate und Ihre technischen Unterlagen vollständig, aktuell und mit der MDR/IVDR konform sind. Freyr sorgt dafür, dass Sie für die Konformitätsbewertung und die Beantragung der CE-Kennzeichnung bestens vorbereitet sind.

Beantwortung von Anfragen zuständiger Behörden
Bei Bedarf übernimmt Freyr in Ihrem Namen die gesamte direkte Kommunikation und die Klärung von Anfragen seitens der zuständigen EU-Behörden oder benannten Stellen, sorgt für zeitnahe und präzise Antworten und trägt dazu bei, Verzögerungen bei Zulassungen oder behördlichen Überprüfungen nach dem Inverkehrbringen zu vermeiden.

Wachsamkeit und Kommunikation bei Vorfällen
Als Ihr EAR fungiert Freyr als primäre Kontaktstelle für sicherheitsrelevante Kommunikation. Bei Bedarf koordinieren wir die Meldung von Zwischenfällen, sicherheitsrelevante Korrekturmaßnahmen vor Ort (FSCA) sowie Vigilanzberichte zwischen dem Hersteller, medizinischen Fachkräften und den Behörden, um eine angemessene Reaktion und die Einhaltung der gesetzlichen Vorschriften sicherzustellen.

Vorbereitung auf Inspektionen und Audits
Freyr verwaltet sämtliche Unterlagen, Korrespondenz und vorgeschriebenen Aufzeichnungen für behördliche Audits und Inspektionen. Unser Team stellt sicher, dass die technische Dokumentation, die Kennzeichnung und die Aufzeichnungen nach dem Inverkehrbringen jederzeit verfügbar sind und den Anforderungen der MDR/IVDR in vollem Umfang entsprechen.
Warum mit Freyr zusammenarbeiten?
- End-to-end Expertise, die von der Zulassung vor der Markteinführung bis hin zur Marktüberwachung nach der Markteinführung reicht und alle Phasen der Compliance abdeckt.
- Eine nachweisliche Erfolgsbilanz mit über 2000 erfolgreich abgeschlossenen Geräteregistrierungen in verschiedenen Kategorien.
- Eine starke EU-Präsenz mit Sitz in Deutschland (EAR), ergänzt durch lokale Regulierungsspezialisten in Reading und unterstützt durch die globalen Umsetzungsteams von Freyr.
- Eine maßgeschneiderte Übergangsplanung, die strategische Unterstützung bietet, um die CE reibungslos und kosteneffizient zu erreichen.
- Lieferteams für mehrere Regionen mit Unterstützung vor Ort durch die in Deutschland ansässigen EU-Niederlassungen von Freyr.
- Nachweisliche Erfolge bei der Anpassung an die MDR/IVDR und der Umstellung auf ältere Medizinprodukte
- Transparente Projektsteuerung mit direkter Kommunikation mit der benannten Stelle

Wir feiern Kundenerfolge
Medizinprodukte
Registrierungs- und LR-Support
Global
Freyr war ein unverzichtbarer Partner, um eine schnelle globale Skalierbarkeit für unser Geschäft mit Software als Medizinprodukt (SaMD) zu erreichen. Als Startup ist der Erwerb von Fachwissen über weltweite Vorschriften mit unerschwinglichen Kosten verbunden. Die wettbewerbsfähigen Preise und maßgeschneiderten Dienstleistungen von Freyr ermöglichten es uns, dieses Fachwissen zu einem Bruchteil der Kosten von Vollzeitkräften zu erhalten. Die Reaktionsfähigkeit und Anpassungsfähigkeit ihres Teams an Projektprioritäten hat unseren Fortschritt erheblich erleichtert. Wir empfehlen Freyr jedem Unternehmen, das fachkundige Beratung und Unterstützung im regulatorischen Bereich für Medizinprodukte sucht.
Arie Henkin
VP – Qualität und Regulierung, mit Sitz in Australien, Führendes SaMD-Unternehmen
Medizinprodukte
Schweizer Repräsentationsdienstleistungen
Japan und Schweiz
Ich genieße die Zusammenarbeit mit Freyr sehr und betrachte sie als einen wirklich wertvollen Gewinn und eine Erweiterung meines eigenen Teams. Sie sind zuverlässig und präzise, und ihre Preise sind wettbewerbsfähig. Darüber hinaus werde ich nicht zögern, wieder mit Freyr zusammenzuarbeiten.
Darren Mansell
Regulatory Affairs Manager bei einem weltweit tätigen Unternehmen für die Entwicklung und Herstellung von Medizinprodukten mit Sitz im Vereinigten Königreich
Medizinprodukte
Registrierungs- und AR-Dienstleistungen
Malaysia und Indonesien
Freyr bietet einen zuverlässigen Service mit Expertise in vielen Ländern. Ich kann mich darauf verlassen, dass Freyr die notwendigen Informationen bereitstellt, um eine fundierte Entscheidung zu treffen, bevor eine formelle Vereinbarung über den Arbeitsumfang getroffen wird. Sobald ein Projekt läuft, agiert das Freyr-Team professionell und führt die Arbeit mit exzellenter Kommunikation über den Fortschritt aus.