
Übersicht zur USFDA Prämarkt-Zulassung für Medizinprodukte
Der USFDA Premarket Approval (PMA)-Prozess ist einer der von der US FDA bereitgestellten Zulassungswege für Medizinprodukte, der primär für FDA-Medizinprodukte der Klasse III konzipiert ist. Der FDA PMA-Zulassungsprozess für Klasse-III-Produkte umfasst sorgfältige wissenschaftliche und regulatorische Bewertungen zur Beurteilung der Sicherheit und Wirksamkeit des Medizinprodukts, um sicherzustellen, dass die höchsten Standards vor der Marktzulassung erfüllt werden.
Vereinbaren Sie einen Termin mit unseren Experten für die Zulassung vor dem Inverkehrbringen
Wer sollte einen Antrag auf USFDA Prämarkt-Zulassung (PMA) für Medizinprodukte einreichen?
Hersteller von Medizinprodukten müssen einen PMA-Antrag einreichen, wenn das Produkt:
- Neuartig ist.
- Einer Hochrisikoklasse angehört.
- Nicht in der Produktdatenbank zur Klassifizierung gefunden werden kann.
- Nicht wesentlich gleichwertig (NSE) zu Produkten der Klassen I, II oder III ist.
Erhalten Sie Expertenberatung für Ihren Antrag auf Marktzulassung
Was ist der Unterschied zwischen 510(k)-, PMA- und De-Novo-Anträgen?
Marktzulassung
- Produkt der Klasse III, das menschliches Leben unterstützt oder ein potenzielles, unangemessenes Risiko für Krankheit oder Verletzung darstellt.
- Das FDA PMA-Zulassungsverfahren erfordert klinische Studien.
- Erfordert eine Vor-Ort-Inspektion, bevor die PMA-Zulassung erteilt wird.
- 180 Kalendertage
De-Novo-Klassifizierung
- Neuartige Produkte der Klassen I und II, die kein gültiges Referenzprodukt haben.
- Erfordert klinische Studiendaten.
- Kein Audit vor Ort vor der De-Novo-Zulassung.
- 150 Kalendertage.
510(k)-Registrierung
- FDA-Medizinprodukte der Klasse III, die eine wesentliche Äquivalenz mit dem Referenzprodukt aufweisen.
- Es sind keine Tests am Menschen erforderlich.
- Kein Audit vor Ort vor der 510(k)-Freigabe.
- 90 Kalendertage.
Welche verschiedenen FDA Prämarkt-Zulassungsantragsmethoden gibt es?
Hersteller können eines der folgenden vier (04) PMA-Antragsverfahren wählen, das am besten für ihr Produkt geeignet ist:
- Traditionelle PMA
- Modulare PMA
- Produktentwicklungsprotokoll
- Ausnahmegenehmigung für humanitäre Produkte
Welche Daten sind für die Marktzulassung von Medizinprodukten erforderlich?
Gemäß 21 CFR Part 814 müssen Antragsteller ein ordnungsgemäß ausgefülltes CDRH-Antragsformular, erforderliche Zusagen und eine gut ausgearbeitete technische PMA-Datei bei der US FDA einreichen. Die technische Datei muss die nicht-klinischen und klinischen Daten enthalten.
Nicht-klinische Daten – Sie umfassen Daten zu Mikrobiologie, Toxikologie, Immunologie, Biokompatibilität, Belastung, Verschleiß, Haltbarkeit sowie andere Labor- oder Tierversuche.
Klinische Daten – Sie umfassen Daten zu Studienprotokollen, Sicherheits- und Wirksamkeitsdaten, unerwünschten Reaktionen und Komplikationen, Geräteausfällen und -ersatz, Patienteninformationen, Patientenbeschwerden, Datentabellen aller einzelnen Probanden, Ergebnissen statistischer Analysen und allen weiteren Informationen aus den klinischen Prüfungen.
Was ist das PMA-Antragsverfahren?
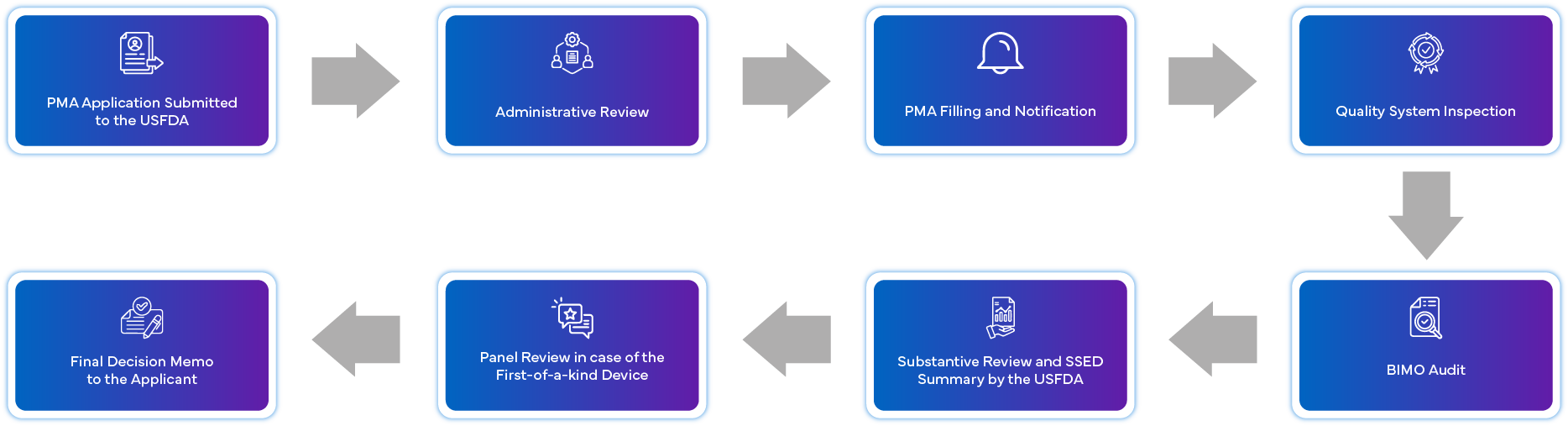
Was sind die Anforderungen an die Compliance nach der Zulassung für PMA?
Die über den PMA-Weg zugelassenen Produkte müssen die von der USFDA festgelegten Anforderungen nach dem Inverkehrbringen erfüllen. Das Produkt muss Folgendes einhalten:
- Anforderungen nach der Zulassung, die in der FDA PMA-Zulassungsanordnung festgelegt sind.
- Management von Änderungen nach der Zulassung durch fristgerechte Einreichung relevanter PMA-Ergänzungen
- Einreichung von Berichten nach der Zulassung (jährlich)
- 21 CFR 803 Vorschriften für die Meldung von Medizinprodukten (MDR)
- Studien zur Überwachung nach dem Inverkehrbringen, wie von der USFDA in den PMA-Zulassungsanordnungen gefordert.
Wie hoch sind die Gebühren der USFDA für die Prüfung des PMA-Antrags?
Die MDUFA-Benutzergebühren für die ursprüngliche PMA und Ergänzungen sind wie folgt:
| Antragstyp | Gebühren für das Geschäftsjahr 2023 (1. Oktober 2022 bis 30. September 2023) | |
|---|---|---|
| Standardgebühr | Gebühr für Kleinunternehmen | |
| PMA, PDP, PMR, BLA | $441,547 |
|
| Panel-Track-Ergänzung | $353,238 | $88,309 |
| 180-Tage-Ergänzung | $66,232 | $16,558 |
| Jahresgebühr für die regelmäßige Berichterstattung über ein Medizinprodukt der Klasse III (PMAs, PDPs und PMRs) | $15,454 | $3,864 |
| 30-Tage-Mitteilung | $7,065 | $3,532 |
| Echtzeit-Ergänzung | $30,908 | $7,727 |
Dank seiner Expertise bei PMA-Einreichungen kann Freyr Sie bei der Ermittlung und Zusammenstellung der erforderlichen Informationen sowie bei der Vorbereitung und Überprüfung des Antrags unterstützen.
Expertise und Vorteile bei der USFDA-Zulassung von Medizinprodukten vor Markteinführung
- Regulatorische Due Diligence
- Konformität bei der Inspektion des Qualitätssystems
- Einhaltung des BIMO-Audits
- Erstellung der PMA-technischen Dokumentation
- Veröffentlichung und Erstellung von eCopy
- Validierung und Einreichung von eCopy
- Bearbeitung von RTA-Antworten und Mängeln
- Vermittlungsdienste bis zur FDA-Zulassung vor Markteinführung
- Beratung bei Mängeln
- Produktlistung und Registrierung der Betriebsstätte
- Verwaltung von PMA-Ergänzungen und 30-Tage-Meldungen
- Jährliche Einreichungen periodischer Berichte
- Scheinaudits und 21 CFR 820 Schulung

- Erfahrung in der Bearbeitung zahlreicher FDA PMA-Einreichungen für verschiedene Gerätekategorien.
- Expertenteam für FDA-Anträge auf Zulassung vor Markteinführung gemäß den regulatorischen Anforderungen
- Zusätzliche Unterstützung zur Bearbeitung von PMA-bezogenen Anfragen.
- Rechtzeitige Einreichung von Leistungen
- Auf dem neuesten Stand der neuen Änderungen der US FDA.
