
Klinischer Bewertungsbericht (CER) für Medizinprodukte Übersicht
Jedes Produkt, das in der Europäischen Union (EU) in Verkehr gebracht werden soll, muss mit einer CE-Kennzeichnung versehen sein. Gemäß der EU MDR variieren die Anforderungen an einen klinischen Bewertungsbericht (CER), einschließlich der Verfahrens- und Datenanforderungen, je nach Produktklasse und sind für die Erlangung der CE-Zertifizierung von Medizinprodukten erforderlich. Produkte der Risikoklasse I können eine CE-Selbstzertifizierung durchführen. Im Gegensatz dazu müssen andere Produktklassen (IIa, IIb, III) die CE-Kennzeichnung durch eine akkreditierte benannte Stelle (NB) vornehmen lassen. Der Hersteller muss die CE-Technische Dokumentation bei der NB zur Bewertung und Erteilung der CE-Kennzeichnung sowie zur Ausstellung des CE-Zertifikats einreichen. Der klinische Bewertungsbericht (CER) für Medizinprodukte muss zusammen mit der CE-Technischen Dokumentation eingereicht werden, um die Anforderungen für die CE-Kennzeichnung zu erfüllen. Der CER sollte kontinuierlich mit neuen Informationen aus Sicherheitsprüfungen, Follow-up-Studien und dem Risikomanagement aktualisiert werden.
Der Klinische Bewertungsbericht (CER) für Medizinprodukte ist einer der Berichte, die zusammen mit der CE-Konformitätsdokumentation zur Erfüllung der CER-Anforderungen eingereicht werden müssen.
Was ist ein Clinical Evaluation Report (CER)?
Die Erstellung von Berichten zur klinischen Bewertung umfasst die Beurteilung und Analyse klinischer Daten eines Medizinprodukts, um dessen klinische Sicherheit und Leistung zu überprüfen. Die klinische Bewertung von Medizinprodukten basiert auf der umfassenden Analyse klinischer Daten vor und nach dem Inverkehrbringen, die für den vorgesehenen Verwendungszweck relevant sind. Der Bericht zur klinischen Bewertung enthält sowohl gerätespezifische Daten als auch Daten zu Produkten, die vom Hersteller als gleichwertig beansprucht werden.
Ein klinischer Bewertungsbericht besteht aus wissenschaftlicher Literatur und analysierten klinischen Daten, die entweder aus einer klinischen Untersuchung Ihres Produkts oder aus den Ergebnissen anderer Studien zu im Wesentlichen gleichwertigen Produkten stammen, wobei die Hersteller uneingeschränkten Zugang zu den technischen, biologischen und klinischen Daten des gleichwertigen Produkts haben müssen und eindeutig nachweisen müssen, inwiefern ihr Produkt in allen entscheidenden Aspekten mit diesem übereinstimmt. Der klinische Bewertungsbericht eines Medizinprodukts belegt, dass das Produkt seinen beabsichtigten Zweck erfüllt, ohne Anwender und Patienten einem zusätzlichen Risiko auszusetzen.
Der EU MDR muss jährlich aktualisiert werden. Die Häufigkeit der Aktualisierungen richtet sich nach dem Risikoprofil und ist produktspezifisch. Wenn das Produkt mit erheblichen Risiken verbunden ist, muss die Aktualisierung mindestens einmal jährlich erfolgen; wenn das Produkt bereits seit längerer Zeit auf dem Markt ist und sich bewährt hat, kann der CER-Bericht alle 2 bis 5 Jahre aktualisiert werden. Jede Änderung am Produktdesign sowie neue Erkenntnisse aus den Daten der Marktüberwachung können eine Aktualisierung des CER-Berichts erforderlich machen.
Die klinische Bewertung von Medizinprodukten, wie im Klinischen Bewertungsbericht (CER) dargelegt, basiert auf den unten aufgeführten Faktoren.
- Derzeit verfügbare wissenschaftliche Literatur; und/oder
- Durchgeführte klinische Prüfungen; oder
- Ob der Nachweis der Konformität mit den grundlegenden Anforderungen auf der Grundlage klinischer Daten nicht als angemessen erachtet wird.
Phasen der Erstellung des Klinischen Bewertungsberichts (CER)
Bezüglich der neuen EU-Medizinprodukte-Verordnung (MDR) – 2017/745 gibt es vier (04) verschiedene Phasen zur Durchführung einer klinischen Bewertung von Medizinprodukten, um einen umfassenden EU-MDR-Bericht zur klinischen Bewertung (CER) zu erstellen.
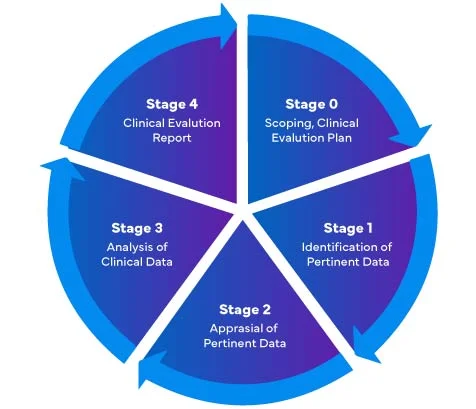
Medizinproduktehersteller, die zum ersten Mal den EU-Markt betreten, müssen sicherstellen, dass ihr Klinischer Bewertungsbericht den EU MDR-Vorschriften entspricht.
Freyr bietet Geräteherstellern End-to-End CE-Zertifizierungsdienstleistungen an, einschließlich der Erstellung von klinischen Bewertungsberichten im Einklang mit den neu implementierten EU MDR 2017/745-Vorschriften. Mit seiner starken regionalen Expertise im Bereich EU-Medizinprodukte wird Freyr den Anforderungen der jeweiligen Behörden gerecht und passt den klinischen Bewertungsbericht entsprechend an.
Klinischer Bewertungsbericht (CER)
- End-to-End Unterstützung bei der Erstellung von klinischen Bewertungsberichten, einschließlich Literaturrecherche, gemäß MEDDEV 2.7/1 Revision 4 und den Leitlinien der EU-Medizinprodukte-Verordnung (MDR).
- Erstellung eines Plans zur klinischen Bewertung für Ihr Unternehmen.
- Die entsprechende anwendbare wissenschaftliche Literatur identifizieren, suchen, analysieren und zusammenstellen.
- Entwicklung einer Vorlage für den Bericht zur klinischen Bewertung für Ihr Unternehmen.
- Lückenanalyse für bestehende Klinische Bewertungsberichte.
- DMS-Tool für Ihr Team, um gemeinsam an der Erstellung von Berichten zur klinischen Bewertung mitzuwirken.
- Integration von PMS-Daten.
- Entwicklung einer Standardarbeitsanweisung für Ihr Team zur Zusammenstellung von PMS-Daten, um Berichte zur klinischen Bewertung zu aktualisieren.
- Bearbeitung regelmäßiger Aktualisierungen bestehender Klinischer Bewertungsberichte gemäß den EU-MDR-Leitlinien.
- PMS-Daten-Unterstützung für bestehende Produkte auf dem Markt.
- CE-Kennzeichnungskonformität und CE-Kennzeichnungsdienstleistungen.

- Gesicherte Einhaltung der neuesten geltenden Vorschriften.
- Team qualifizierter klinischer Experten.
- Funktionsübergreifende Beiträge von Medizinprodukteexperten zur Einhaltung der Anforderungen.
- Umfassender Service, der Compliance, Überprüfung und Planung abdeckt.

Häufig gestellte Fragen (FAQs)
01. Was ist ein klinischer Bewertungsbericht (CER) und warum ist er wichtig?
Ein klinischer Bewertungsbericht (CER) ist ein wissenschaftliches Zulassungsdokument, in dem alle verfügbaren klinischen Nachweise systematisch ausgewertet werden, um nachzuweisen, dass ein Medizinprodukt sicher ist, die beabsichtigte Leistung erbringt und für den vorgesehenen Verwendungszweck gemäß EU MDR ein akzeptables Nutzen-Risiko-Verhältnis aufweist. Er spielt eine zentrale Rolle bei der Konformitätsbewertung und der laufenden Einhaltung der Vorschriften.
02. Wann sollte ein CER erstellt und aktualisiert werden?
Die Erstellung der klinischen Bewertung beginnt bereits in einer frühen Phase der Produktentwicklung als Teil eines geplanten klinischen Bewertungsprozesses und muss während des gesamten Lebenszyklus des Produkts regelmäßig aktualisiert werden, sobald neue klinische Erkenntnisse, Daten aus der Zeit nach der Markteinführung oder Änderungen des Risikoprofils vorliegen, um sicherzustellen, dass die Nutzen-Risiko-Bewertung auf dem neuesten Stand bleibt.
03. Welche wesentlichen Bestandteile muss ein konformer CER enthalten?
Ein fundiertes CER sollte eine strukturierte Bewertung der relevanten klinischen Daten, Vergleiche mit dem aktuellen Stand der Technik, eine Nutzen-Risiko-Analyse, Erkenntnisse aus der Marktüberwachung sowie objektive Schlussfolgerungen zur Übereinstimmung mit EU MDR und Leistungsanforderungen EU MDR widerspiegeln.
04. Wie wirkt sich der „Stand der Technik“ auf einen klinischen Bewertungsbericht aus?
„Stand der Technik“ ist der Maßstab für die anerkannte klinische Praxis und Technologie, an dem klinische Evidenz gemessen wird; er stellt sicher, dass Nutzen und Risiken des Produkts im Kontext der aktuellen medizinischen Standards betrachtet werden, und dient als Leitfaden für eine aussagekräftige Interpretation der Evidenz.
05. Ist gemäß EU MDR für alle Medizinprodukte eine Konformitätserklärung erforderlich?
Ja, CERs sind für alle Medizinprodukte, die gemäß der MDR in der EU in Verkehr gebracht werden, unabhängig von der Risikoklasse verpflichtend, da sie klinische Nachweise dokumentieren, die für den Nachweis der Konformität mit den regulatorischen Sicherheits- und Leistungsanforderungen unerlässlich sind.
06. Was unterscheidet einen hochwertigen CER von einem einfachen Compliance-Bericht?
Ein hochwertiger CER vereint eine umfassende Methodik der Literaturrecherche, klare klinische Aussagen, die auf den Verwendungszweck abgestimmt sind, eine strenge Datenauswertung sowie fundierte Erkenntnisse zu klinischer Wirksamkeit und Risiken. Er geht über das bloße Abhaken von Kriterien hinaus und spiegelt ein tiefgreifendes Verständnis der regulatorischen Erwartungen und des klinischen Kontexts wider.
07. Warum gilt Freyr als führender Anbieter von Dienstleistungen im Bereich klinischer Bewertungsberichte (CER)?
Freyr ist weithin für seinen „Regulatory-First“-Ansatz bei der Entwicklung von CERs bekannt, der fundiertes EU MDR , eine solide Bewertung klinischer Evidenz und lebenszyklusorientierte Strategien vereint. Die multidisziplinären Teams des Unternehmens bringen klinische, regulatorische und postmarktbezogene Erkenntnisse in Einklang, um wissenschaftlich fundierte CERs zu erstellen, die der Prüfung durch benannte Stellen standhalten und gleichzeitig die langfristige Compliance gewährleisten.







