


Übersicht zur Registrierung von Medizinprodukten in der Türkei
Der türkische Markt für Medizinprodukte hat in den letzten zehn Jahren ein erhebliches und konstantes Wachstum verzeichnet. Ab 2021 muss die Registrierung von Medizinprodukten in der Türkei die EU-Verordnungen Medizinprodukte-Verordnung (MDR) 2017/745 und In-vitro-Diagnostika-Verordnung (IVDR) 2017/746 einhalten. Dies hat den internationalen Handel gefördert und dazu geführt, dass mehrere globale Unternehmen ihre Medizinprodukte in dem Land auf den Markt bringen.
![]()
Aufsichtsbehörde: Türkische Arzneimittel- und Medizinprodukteagentur (TITCK)![]()
Verordnung: Medizinprodukte-Verordnung (MDR) 2017/745, In-vitro-Diagnostika-Verordnung 2017/746![]()
Regulatorischer Weg: Die CE-Kennzeichnung ist obligatorisch, gefolgt von der Registrierung/Meldung im Produktverfolgungssystem (UTS)![]()
Lokaler Vertreter in der Türkei![]()
QMS-Anforderung: ISO 13485:2016![]()
Bewertung technischer Daten: Benannte Stelle für die CE-Kennzeichnung![]()
Gültigkeit der Lizenz: Unbegrenzt![]()
Einreichungsformat: Papier![]()
Übersetzung: Übersetzte Dokumente auf Türkisch
Geräteklassifizierung
Die Türkei wendet dieselbe Klassifizierung für Medizinprodukte an, wie sie in der EU MDR und IVDR festgelegt ist. Die Bestimmung der Produktklassifizierung kann eine Herausforderung sein. Daher ist die Unterstützung durch einen erfahrenen Berater für regulatorische Angelegenheiten hier sehr wichtig.
Medizinprodukteklassen
| Klasse | Risiko |
|---|---|
| Klasse I | Niedrig |
| Klasse IIa | Mittel |
| Klasse IIb | Mittel bis Hoch |
| Klasse III | Hoch |
Klassen von In-vitro-Diagnostika
| Klasse | Risiko |
|---|---|
| Klasse A | Niedrig |
| Klasse B | Mittel |
| Klasse C | Mittel bis Hoch |
| Klasse D | Hoch |
Registrierung von Medizinprodukten
Die CE-Kennzeichnung ist eine Konformitätsanforderung, die Hersteller erfüllen müssen, um ihre Produkte auf dem türkischen Markt zu platzieren. Sie wird nach einer Konformitätsbewertung durch eine benannte Stelle erteilt. Die Türkei ist nun berechtigt, benannte Stellen gemäß EU MDR und IVDR zu ernennen.
Die Unternehmen müssen sich im Zentralen Registrierungssystem (MERSIS) registrieren und das Medizinprodukt im Produktverfolgungssystem (UTS) registrieren.
Prozessablauf
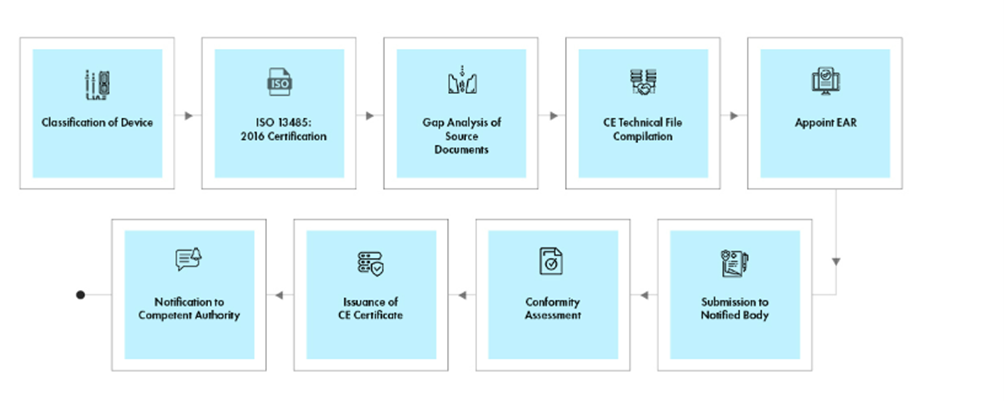
Lebenszyklusmanagement von Medizinprodukten nach der Zulassung
Freyr unterstützt ausländische Hersteller beim End-to-End-Lebenszyklusmanagement von Medizinprodukten, einschließlich Aktivitäten nach der Zulassung, wie zum Beispiel:
- Änderungsmanagement nach der Zulassung – Änderungen an bestehenden Medizinproduktezulassungen, wie die Hinzufügung neuer Varianten, Zubehörteile; die Hinzufügung neuer Anwendungsindikationen und Ähnliches.
- Aufrechterhaltung der ISO 13485:2016 und CE-Zertifizierung
- Verlängerung von Lizenzen
- Koordination zwischen Benannter Stelle und Hersteller
Da verschiedene Genehmigungsstellen beteiligt sind, müssen ausländische Hersteller in jedem einzelnen Prozess für Gerätegenehmigungen mehrere Vorschriften einhalten. Die Erlangung einer CE-Kennzeichnung und die weitere Einhaltung landesspezifischer Vorschriften erfordert umfassendes regulatorisches Wissen. Manchmal kann es für Markteinsteiger ohne einen bewährten regulatorischen Partner schwierig sein, alle Geräteanforderungen zu erfüllen. Um Hersteller zu unterstützen, bietet Freyr End-to-End Regulatory Services an, um die Zulassungen für Medizinprodukte zu beschleunigen.
Freyr Expertise
- Europäische Klassifizierung von Medizinprodukten
- Unterstützung durch einen Europäischen Bevollmächtigten (EAR)
- Registrierung von Medizinprodukten und Produktmeldung in der Türkei
- ISO 14971:2019 Beratung zum Risikomanagement
- ISO 13485:2016 Konformität
- Überprüfung, Erstellung und Einreichung von CE-Konformitätsdokumentation/Design-Dossier
- EU MDR Übergangsunterstützung
- Unterstützung beim Übergang zur EU IVDR
- Klinische Bewertungsberichte (CER) für Medizinprodukte
- Leistungsbewertungsberichte (PER) für In-vitro-Diagnostika
- Meldung/Registrierung von Medizinprodukten über ein Online-Registrierungssystem
- Regulatorischer Strategiebericht für Medizinprodukte
- Testunterstützung – Biokompatibilität, elektrische Sicherheit, mechanische Eigenschaften und Leistung
- Unterstützung bei der Einhaltung der Kennzeichnungsvorschriften
- GMP-Unterstützung
- Unterstützung bei der Post-Market-Überwachung
