


Przegląd rejestracji wyrobów medycznych w Turcji
Turecki rynek wyrobów medycznych odnotował znaczący i stały wzrost w ciągu ostatniej dekady. Od 2021 roku rejestracja wyrobów medycznych w Turcji wymaga przestrzegania Rozporządzenia UE w sprawie wyrobów medycznych (MDR) 2017/745 oraz Rozporządzenia w sprawie wyrobów medycznych do diagnostyki in vitro (IVDR) 2017/746. Wzmocniło to handel międzynarodowy, co skłoniło wiele globalnych firm do wprowadzania swoich wyrobów medycznych w tym kraju.
![]()
Organ regulacyjny: Turecka Agencja Leków i Wyrobów Medycznych (TITCK)![]()
Regulacja: Rozporządzenie w sprawie wyrobów medycznych (MDR) 2017/745, Rozporządzenie w sprawie wyrobów medycznych do diagnostyki in vitro 2017/746![]()
Ścieżka regulacyjna: Oznakowanie CE jest obowiązkowe, a następnie rejestracja/zgłoszenie w Systemie Śledzenia Produktów (UTS).![]()
Lokalny przedstawiciel w Turcji![]()
Wymóg QMS: ISO 13485:2016![]()
Ocena Danych Technicznych: Jednostka notyfikowana do oznakowania CE![]()
Ważność licencji: Nieograniczone![]()
Format przedłożenia: Papier![]()
Tłumaczenie: Przetłumaczone dokumenty na język turecki
Klasyfikacja wyrobów
Turcja stosuje tę samą klasyfikację wyrobów medycznych, co określono w EU MDR i IVDR. Ustalenie klasyfikacji wyrobu może być wyzwaniem, dlatego wsparcie doświadczonego konsultanta regulacyjnego jest tutaj kluczowe.
Klasy wyrobów medycznych
| Klasa | Ryzyko |
|---|---|
| Klasa I | Niski |
| Klasa IIa | Umiarkowany |
| Klasa IIb | Od umiarkowanego do wysokiego |
| Klasa III | Wysoki |
Klasy wyrobów medycznych do diagnostyki in vitro
| Klasa | Ryzyko |
|---|---|
| Klasa A | Niski |
| Klasa B | Umiarkowany |
| Klasa C | Od umiarkowanego do wysokiego |
| Klasa D | Wysoki |
Rejestracja wyrobów medycznych
Oznakowanie CE to potwierdzenie zgodności, które jest wymagane od producentów, aby mogli wprowadzić swoje urządzenie na rynek turecki. Oznakowanie CE jest wydawane w drodze oceny zgodności przeprowadzanej przez jednostkę notyfikowaną. Obecnie Turcja jest uprawniona do wyznaczania jednostek notyfikowanych zgodnie z EU MDR i IVDR.
Firmy są zobowiązane do rejestracji w Centralnym Systemie Rejestracji (MERSIS) oraz zarejestrowania wyrobu w Systemie Śledzenia Produktów (UTS).
Przebieg procesu
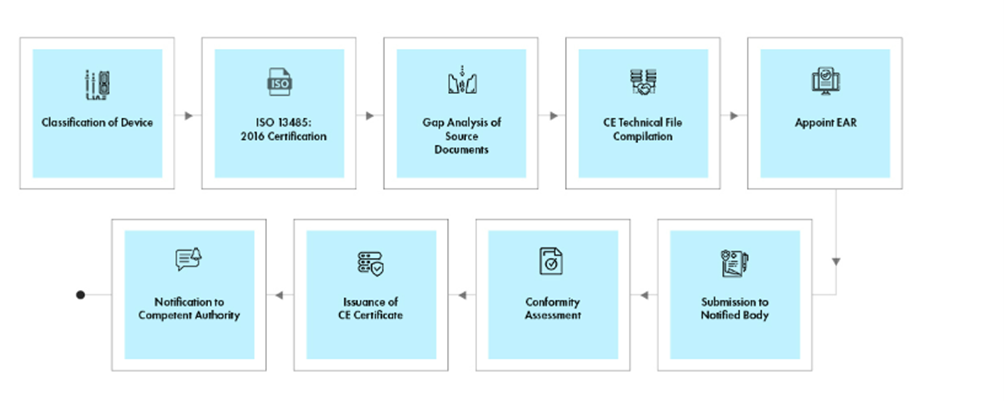
Zarządzanie cyklem życia wyrobu po zatwierdzeniu
Freyr wspiera zagranicznych producentów w zarządzaniu cyklem życia wyrobów medycznych End-to-End, w tym w działaniach po zatwierdzeniu, takich jak:
- Zarządzanie zmianami po zatwierdzeniu – modyfikacje istniejących zatwierdzeń wyrobów medycznych, takie jak dodawanie nowych wariantów, akcesoriów; dodawanie nowych wskazań do stosowania i inne
- Utrzymywanie certyfikacji ISO 13485:2016 i CE
- Odnowienie licencji
- Pośrednictwo między jednostką notyfikowaną a producentem
W związku z zaangażowaniem różnych organów autoryzacyjnych, zagraniczni producenci muszą przestrzegać wielu zestawów przepisów w każdym indywidualnym procesie zatwierdzania wyrobów. Uzyskanie oznakowania CE i dalsze przestrzeganie przepisów obowiązujących w poszczególnych stanach wymaga rozległej wiedzy regulacyjnej. Czasami, bez sprawdzonego partnera regulacyjnego, poruszanie się po wszystkich wymaganiach dotyczących wyrobów może być wyzwaniem dla nowych uczestników rynku. Aby pomóc producentom, Freyr świadczy usługi regulacyjne End-to-End w celu przyspieszenia zatwierdzeń dla wyrobów medycznych.
Ekspertyza Freyr
- Europejska Klasyfikacja Wyrobów Medycznych
- Wsparcie Europejskiego Upoważnionego Przedstawiciela (EAR)
- Rejestracja urządzeń i zgłaszanie produktów w Turcji
- ISO 14971:2019 Konsultacje w zakresie zarządzania ryzykiem
- Zgodność z ISO 13485:2016
- Przegląd, opracowanie i złożenie dokumentacji technicznej CE/dossier projektowego.
- Wsparcie w przejściu na EU MDR
- Wsparcie w przejściu na unijne rozporządzenie IVDR
- Clinical Evaluation Reports (CER) wyroby medyczne
- Raporty z oceny wydajności (PER) dla wyrobów medycznych do diagnostyki in vitro
- Powiadamianie/Rejestracja wyrobów medycznych za pośrednictwem internetowego systemu rejestracji
- Raport ze strategii regulacyjnej dla wyrobów medycznych
- Wsparcie w testowaniu – biokompatybilność, bezpieczeństwo elektryczne, mechanika i wydajność
- Wsparcie w zakresie zgodności oznakowania
- Wsparcie GMP
- Wsparcie w zakresie nadzoru po wprowadzeniu do obrotu
