
Raport z oceny klinicznej (CER) dla wyrobów medycznych Przegląd
Każde wyroby medyczne przeznaczone do wprowadzenia do obrotu w Unii Europejskiej (UE) muszą posiadać oznaczenie CE. Zgodnie z rozporządzeniem EU MDR wymagania dotyczące raportu z oceny klinicznej (CER), w tym wymagania dotyczące procesu i danych, różnią się w zależności od klasy wyrobu i są niezbędne do uzyskania certyfikatu CE dla wyroby medyczne. W przypadku wyrobów medycznych klasy I o niskim ryzyku można przeprowadzić samocertyfikację CE. Natomiast w przypadku innych klas wyrobów medycznych (IIa, IIb, III) certyfikacja znaku CE musi zostać przeprowadzona przez akredytowaną jednostkę notyfikowaną (NB). Producent musi przedłożyć dokumentację techniczną CE jednostce notyfikowanej w celu oceny i wydania zgody na znak CE oraz wydania certyfikatu CE. Raport z oceny klinicznej (CER) dla wyroby medyczne przedłożyć wraz z dokumentacją techniczną CE, aby spełnić wymagania dotyczące oznakowania CE. Raport CER powinien być na bieżąco aktualizowany o nowe informacje pochodzące z kontroli bezpieczeństwa, badań uzupełniających i zarządzania ryzykiem.
Raport z oceny klinicznej (CER) dla wyrobów medycznych jest jednym z raportów, które należy złożyć wraz z Dokumentacją techniczną CE w celu spełnienia wymagań CER.
Czym jest Raport z Oceny Klinicznej (CER)?
Sporządzanie raportu z oceny klinicznej obejmuje ocenę i analizę danych klinicznych dotyczących wyrobu medycznego w celu weryfikacji jego bezpieczeństwa klinicznego i działania. Ocena kliniczna wyrobów medycznych opiera się na kompleksowej analizie danych klinicznych przed- i powprowadzeniowych, istotnych dla zamierzonego zastosowania. Raport z oceny klinicznej zawiera dane specyficzne dla wyrobu, a także wszelkie dane dotyczące wyrobów uznanych przez producenta za równoważne.
Raport z oceny klinicznej składa się z literatury naukowej oraz przeanalizowanych danych klinicznych zebranych albo w ramach badań klinicznych danego wyrobu, albo na podstawie wyników innych badań dotyczących wyrobów zasadniczo równoważnych, przy czym producenci muszą mieć pełny dostęp do danych technicznych, biologicznych i klinicznych dotyczących wyrobu równoważnego oraz jasno wykazać, w jaki sposób ich wyrób odpowiada mu pod każdym istotnym względem. Raport z oceny klinicznej wyrobu medycznego potwierdza, że wyrób ten spełnia swoje przeznaczenie bez narażania użytkowników i pacjentów na dodatkowe ryzyko.
EU MDR musi być aktualizowany co roku. Częstotliwość aktualizacji raportu CER zależy od poziomu ryzyka i jest uzależniona od konkretnego wyrobu. Jeśli wyrób wiąże się ze znacznym ryzykiem, aktualizację należy przeprowadzać co najmniej raz w roku; natomiast w przypadku gdy wyrób jest wprowadzany do obrotu od dłuższego czasu i ma ugruntowaną pozycję na rynku, raport CER można aktualizować co 2–5 lat. Wszelkie zmiany w konstrukcji wyrobu oraz nowe informacje pochodzące z danych z monitorowania po wprowadzeniu do obrotu mogą stanowić podstawę do aktualizacji raportu CER.
Ocena kliniczna wyrobów medycznych, przedstawiona w raporcie z oceny klinicznej (CER), opiera się na czynnikach wymienionych poniżej.
- Dostępna literatura naukowa; i/lub
- przeprowadzone badania kliniczne; lub
- W przypadku, gdy wykazanie zgodności z zasadniczymi wymaganiami na podstawie danych klinicznych nie jest uznane za stosowne.
Etapy pisania Raportu Oceny Klinicznej (CER)
Odnosząc się do nowego rozporządzenia UE w sprawie wyrobów medycznych (MDR) – 2017/745, istnieją cztery (04) różne etapy przeprowadzania oceny klinicznej wyrobów medycznych w celu przygotowania kompleksowego Raportu z Oceny Klinicznej (CER) zgodnego z unijnym rozporządzeniem MDR.
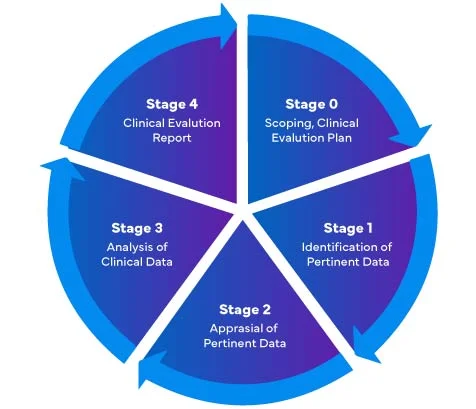
Producenci wyrobów medycznych wchodzący po raz pierwszy na rynek UE muszą upewnić się, że ich Raport z Oceny Klinicznej jest zgodny z przepisami EU MDR.
Freyr świadczy kompleksowe usługi certyfikacji CE End-to-End dla producentów wyrobów, w tym sporządzanie Raportów Oceny Klinicznej zgodnie z nowo wdrożonymi przepisami EU MDR 2017/745. Dzięki silnemu regionalnemu doświadczeniu w zakresie wyrobów medycznych w UE, Freyr dostosowuje się do wymagań poszczególnych agencji i odpowiednio dostosowuje Raport Oceny Klinicznej.
Raport z oceny klinicznej (CER)
- End-to-end wsparcie w pisaniu Raportu Oceny Klinicznej, w tym wyszukiwanie literatury, zgodnie z MEDDEV 2.7/1 rewizja 4 oraz wytycznymi Rozporządzenia UE w sprawie wyrobów medycznych (MDR).
- Opracowywanie planu oceny klinicznej dla Państwa organizacji.
- Zidentyfikować, wyszukać, przeanalizować i zebrać odpowiednią, mającą zastosowanie literaturę naukową.
- Opracuj szablon Raportu z Oceny Klinicznej dla Twojej organizacji.
- Analiza luk dla istniejącego raportu z oceny klinicznej.
- Narzędzie DMS dla Twojego zespołu do wspólnego tworzenia raportów z oceny klinicznej.
- Integrowanie danych z PMS.
- Opracuj standardową procedurę operacyjną dla Twojego zespołu do gromadzenia danych PMS w celu aktualizacji Raportów z Oceny Klinicznej.
- Obsługa okresowych aktualizacji istniejących raportów z oceny klinicznej, zgodnie z wytycznymi EU MDR.
- Wsparcie danymi PMS dla istniejących wyrobów na rynku.
- Zgodność z oznakowaniem CE oraz usługi w zakresie oznakowania CE.

- Zapewniona zgodność z najnowszymi obowiązującymi przepisami.
- Zespół wykwalifikowanych ekspertów klinicznych.
- Wkład interdyscyplinarny od ekspertów ds. wyrobów medycznych w celu spełnienia wymagań.
- Kompleksowa usługa w zakresie zgodności, przeglądu i planowania.

Często Zadawane Pytania (FAQ)
01. Czym jest raport z oceny klinicznej (CER) i dlaczego ma on znaczenie?
Raport z oceny klinicznej (CER) to naukowy dokument regulacyjny, w którym w sposób systematyczny dokonuje się oceny wszystkich dostępnych dowodów klinicznych w celu wykazania, że wyrób medyczny jest bezpieczny, działa zgodnie z przeznaczeniem oraz zapewnia akceptowalny stosunek korzyści klinicznych do ryzyka w ramach przeznaczenia określonego w EU MDR. Odgrywa on kluczową rolę w ocenie zgodności oraz w zapewnianiu stałej zgodności z przepisami.
02. Kiedy należy sporządzić i zaktualizować certyfikat zgodności (CER)?
Przygotowanie dokumentacji klinicznej rozpoczyna się na wczesnym etapie opracowywania produktu w ramach zaplanowanego procesu oceny klinicznej i musi być regularnie aktualizowane przez cały cykl życia wyrobu, ilekroć pojawiają się nowe dowody kliniczne, dane po wprowadzeniu do obrotu lub zmiany w profilu ryzyka, co gwarantuje, że ocena stosunku korzyści do ryzyka pozostaje aktualna.
03. Jakie podstawowe elementy musi zawierać certyfikat CER zgodny z wymogami?
Solidna ocena zgodności powinna odzwierciedlać uporządkowaną analizę odpowiednich danych klinicznych, porównania z najnowszym stanem wiedzy, analizę stosunku korzyści do ryzyka, wyniki nadzoru po wprowadzeniu do obrotu oraz obiektywne wnioski dotyczące zgodności z wymogami EU MDR i działania określonymi w EU MDR .
04. W jaki sposób „najnowszy stan wiedzy” wpływa na raport z oceny klinicznej?
„Najnowszy stan wiedzy” stanowi punkt odniesienia dla uznanej praktyki klinicznej i technologii, z którym porównuje się dane kliniczne; gwarantuje on, że korzyści i ryzyko związane z urządzeniem są rozpatrywane w kontekście aktualnych standardów medycznych, co pozwala na trafną interpretację wyników badań.
05. Czy wyroby medyczne EU MDR certyfikat zgodności (CER) jest wymagany dla wszystkich wyroby medyczne ?
Tak, certyfikaty CER są obowiązkowe dla wszystkich wyroby medyczne w UE zgodnie z rozporządzeniem MDR, niezależnie od klasy ryzyka, ponieważ dokumentują one dowody kliniczne niezbędne do wykazania zgodności z regulacyjnymi wymogami dotyczącymi bezpieczeństwa i działania.
06. Czym różni się wysokiej jakości raport CER od podstawowego raportu zgodności?
Wysokiej jakości dokumentacja kliniczna (CER) łączy w sobie kompleksową metodologię przeglądu literatury, jasno sformułowane twierdzenia kliniczne dostosowane do przeznaczenia produktu, rygorystyczną ocenę danych oraz przemyślane spostrzeżenia dotyczące skuteczności klinicznej i ryzyka, wykraczając poza rutynowe wypełnianie formularzy i odzwierciedlając dogłębne zrozumienie oczekiwań organów regulacyjnych oraz kontekstu klinicznego.
07. Dlaczego firma Freyr jest uznawana za wiodącego partnera w zakresie usług związanych z raportami z badań klinicznych (CER)?
Firma Freyr cieszy się powszechnym uznaniem dzięki podejściu do opracowywania badań porównawczych opartych na dowodach naukowych (CER), w którym na pierwszym miejscu stawia kwestie regulacyjne, łącząc dogłębną EU MDR , rzetelną ocenę dowodów klinicznych oraz strategie ukierunkowane na cały cykl życia produktu. Jej multidyscyplinarne zespoły łączą wiedzę z zakresu kliniki, regulacji prawnych i działań po wprowadzeniu produktu do obrotu, aby dostarczać rygorystyczne naukowo badania CER, które wytrzymują kontrolę jednostek notyfikowanych, a jednocześnie zapewniają długoterminową zgodność z przepisami.







