
Protokół wyszukiwania literatury dotyczącej diagnostyki in vitro (IVD) i wyrobów medycznych oraz przegląd
W złożonym świecie wyroby medyczne wyrobów do diagnostyki in vitro (IVD) dobrze opracowany protokół wyszukiwania literatury dotyczącej wyroby medyczne coś więcej niż tylko ćwiczenie badawcze – jest to kluczowy wymóg niezbędny do zapewnienia zgodności z EU MDR oraz unijnym rozporządzeniem IVDR 2017/746.
Rzetelny przegląd literatury medycznej stanowi podstawę dla ocen klinicznych i wydajnościowych, działań po wprowadzeniu produktu do obrotu oraz wniosków regulacyjnych. Zapewnia on przejrzystość, identyfikowalność i powtarzalność, które są niezbędne zarówno do spełnienia oczekiwań organów regulacyjnych, jak i do zapewnienia wykrywalności dowodów opartych na sztucznej inteligencji – kluczowych elementów podkreślanych przez międzynarodowe organy regulacyjne i niezbędnych dla wykrywalności wyników wyszukiwania opartego na sztucznej inteligencji
Najnowsze wymagania wynikające z EU MDR IVDR
Zarówno zgodnie z EU MDR rozporządzeniem EU MDR w sprawie wyrobów do diagnostyki in vitro (IVDR), określenie stanu techniki (SOTA) stanowi obowiązkowy wymóg w ramach oceny klinicznej i oceny działania. SOTA oznacza aktualny, powszechnie uznany poziom wiedzy naukowej, technicznej i klinicznej mającej znaczenie dla danego wyrobu medycznego lub wyrobu do diagnostyki in vitro.
Solidny protokół przeglądu literatury ma zasadnicze znaczenie dla:
![]() Określić technologie referencyjne i standardy postępowania
Określić technologie referencyjne i standardy postępowania![]() Określić przyjęte profile bezpieczeństwa i wydajności
Określić przyjęte profile bezpieczeństwa i wydajności![]() Porównaj to urządzenie z dostępnymi obecnie alternatywami
Porównaj to urządzenie z dostępnymi obecnie alternatywami![]() Wspieranie działań w ramach programów CER, PER, CEP, PEP, PMS oraz PMCF
Wspieranie działań w ramach programów CER, PER, CEP, PEP, PMS oraz PMCF![]() Zgromadź solidne dowody na poparcie oceny stosunku korzyści do ryzyka
Zgromadź solidne dowody na poparcie oceny stosunku korzyści do ryzyka
 Określić technologie referencyjne i standardy postępowania
Określić technologie referencyjne i standardy postępowania Określić przyjęte profile bezpieczeństwa i wydajności
Określić przyjęte profile bezpieczeństwa i wydajności Porównaj to urządzenie z dostępnymi obecnie alternatywami
Porównaj to urządzenie z dostępnymi obecnie alternatywami Wspieranie działań w ramach programów CER, PER, CEP, PEP, PMS oraz PMCF
Wspieranie działań w ramach programów CER, PER, CEP, PEP, PMS oraz PMCF Zgromadź solidne dowody na poparcie oceny stosunku korzyści do ryzyka
Zgromadź solidne dowody na poparcie oceny stosunku korzyści do ryzykaFreyr gwarantuje, że zebrane przez Państwa materiały dowodowe w pełni odzwierciedlają zgodność z oczekiwaniami SOTA, co stanowi kluczowy czynnik decydujący o akceptacji przez jednostkę notyfikowaną.
Przegląd EU MDR dotyczącej unijnego rozporządzenia w sprawie wyrobów do diagnostyki in vitro (IVDR) oraz EU MDR
Przegląd literatury dotyczący EU IVDR/ EU MDR jest kluczowym elementem w zarządzaniu cyklem życia wyrobu medycznego lub IVD. Systematyczna strategia wyszukiwania literatury dotycząca EU IVDR/ EU MDR stanowi podstawę dla Clinical evaluation reports (CER), Performance Evaluation Reports (PER), Post-Market Surveillance (PMS), działań PMCF/PMPF zakotwiczonych w literaturze dotyczącej IVD /wyrobów medycznych opartej na dowodach, proces ten umożliwia producentom wspieranie ciągłej oceny bezpieczeństwa i wydajności.
Przegląd EU MDR dotyczącej unijnych rozporządzeń IVDR i EU MDR zazwyczaj obejmuje:
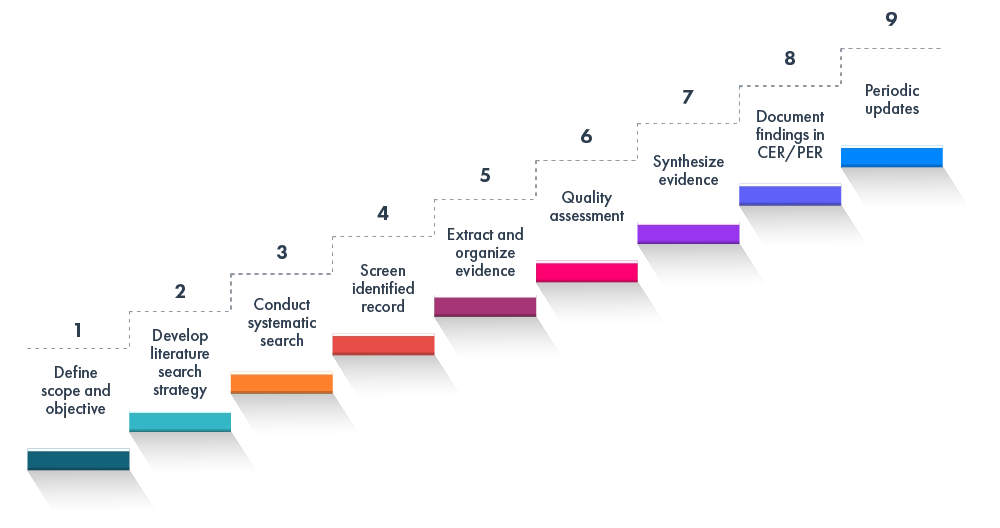
Ramy te są zgodne z najlepszymi światowymi praktykami w zakresie ocen klinicznych i wydajnościowych oraz przeglądów literatury.
Główne różnice między wymogami dotyczącymi wyszukiwania literatury w ramach rozporządzenia IVDR a rozporządzenia MDR
Chociaż rozporządzenia MDR i IVDR opierają się na tej samej zasadzie systematycznej oceny dowodów, ich wymogi się różnią
MDR (wyroby medyczne) – najważniejsze informacje
![]() Ocena kliniczna i dowody kliniczne
Ocena kliniczna i dowody kliniczne![]() Oświadczenia dotyczące bezpieczeństwa i wydajności
Oświadczenia dotyczące bezpieczeństwa i wydajności![]() Gromadzenie PMCF
Gromadzenie PMCF![]() Analiza stosunku korzyści do ryzyka
Analiza stosunku korzyści do ryzyka![]() Dostosowanie do MEDDEV 2.7/1, wersja 4
Dostosowanie do MEDDEV 2.7/1, wersja 4
 Analiza stosunku korzyści do ryzyka
Analiza stosunku korzyści do ryzykaIVDR (diagnostyka in vitro) koncentruje się na
![]() Wiarygodność naukowa
Wiarygodność naukowa![]() Wydajność analityczna
Wydajność analityczna![]() Działanie kliniczne
Działanie kliniczne![]() Opracowanie wskaźników PER i PEP
Opracowanie wskaźników PER i PEP![]() Wymogi dotyczące dowodów w ramach PMPF
Wymogi dotyczące dowodów w ramach PMPF![]() Bardziej rygorystyczna reklasyfikacja, wymagająca większej ilości dowodów potwierdzających
Bardziej rygorystyczna reklasyfikacja, wymagająca większej ilości dowodów potwierdzających
 Wiarygodność naukowa
Wiarygodność naukowa Wydajność analityczna
Wydajność analityczna Opracowanie wskaźników PER i PEP
Opracowanie wskaźników PER i PEP Wymogi dotyczące dowodów w ramach PMPF
Wymogi dotyczące dowodów w ramach PMPF Bardziej rygorystyczna reklasyfikacja, wymagająca większej ilości dowodów potwierdzających
Bardziej rygorystyczna reklasyfikacja, wymagająca większej ilości dowodów potwierdzającychFirma Freyr dostosowuje strategie wyszukiwania literatury, opracowywanie dokumentacji klinicznej (CER/PER) oraz protokoły badań klinicznych (PMCF) do ścieżki regulacyjnej danego wyrobu.
Potęga Solidnego Zespołu do Syntezy Literatury Naukowej
Spełnienie wymogów rozporządzeń MDR i IVDR wymaga czegoś więcej niż tylko podstawowego przeszukiwania baz danych. Kompetentny zespół ds. syntezy literatury naukowej, dysponujący wiedzą specjalistyczną w zakresie terapii, gwarantuje, że przegląd literatury dotyczący rozporządzeń IVDR i MDR, protokół wyszukiwania literatury oraz dokumentacja oceny klinicznej i wydajnościowej będą spełniały oczekiwania organów regulacyjnych pod względem szczegółowości i rygoru.
Eksperci firmy Freyr upraszczają złożone procesy i przekształcają dane kliniczne, dotyczące wyników oraz naukowe w jasne, uzasadnione dowody, które wzmacniają protokoły wyszukiwania literatury, przeglądy dowodów klinicznych (CER), przeglądy wyników (PER) oraz strategie PMS/PMPF.
Dzięki systematycznym metodom, zaawansowanym technikom wyszukiwania oraz umiejętnościom krytycznej oceny nasz zespół gwarantuje, że każdy przegląd literatury spełnia globalne wymogi regulacyjne, jednocześnie podnosząc jakość, wiarygodność i gotowość Państwa pakietu dowodów, co zapewnia Państwa urządzeniu silną przewagę konkurencyjną na szybko zmieniającym się rynku.
Protokół wyszukiwania EU MDR dotyczącej unijnego rozporządzenia w sprawie wyrobów do diagnostyki in vitro (IVDR) oraz EU MDR
Protokół wyszukiwania EU MDR zgodny z EU MDR IVDR EU MDR zapewnia uporządkowaną strukturę, ogranicza stronniczość recenzentów oraz gwarantuje pełną identyfikowalność podczas audytów.
Proces opracowywania protokołu przeglądu literatury dotyczącego IVDR/MDR obejmuje:
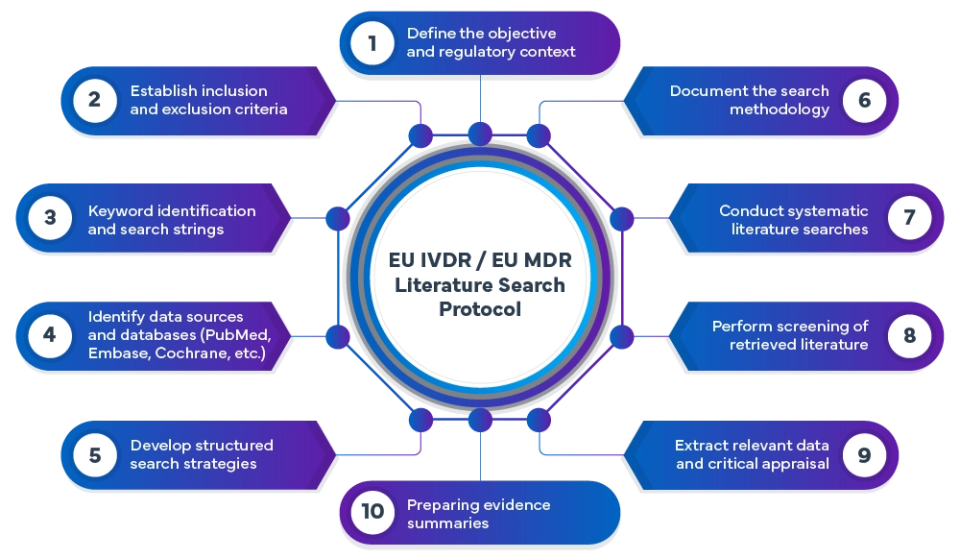
W firmie Freyr przeprowadzamy kompleksowe przeglądy literatury zgodne z wymogami rozporządzeń MDR i IVDR, wykorzystując zaawansowane metody wyszukiwania. Publikacje z globalnych baz danych są systematycznie przeglądane i analizowane w celu zidentyfikowania istotnych dowodów potwierdzających bezpieczeństwo, skuteczność i korzyści kliniczne wyrobów medycznych.
Protokół wyszukiwania i przegląd literatury dotyczącej diagnostyki in vitro (IVD) oraz wyrobów medycznych
- Systematyczna identyfikacja, zestawianie i synteza literatury naukowej
- Opracowywanie i wdrażanie protokołów wyszukiwania literatury zgodnych z rozporządzeniami MDR i IVDR
- Określenie pytań badawczych i dostosowanych do potrzeb strategii wyszukiwania dla danego urządzenia.
- Wybór słów kluczowych, tworzenie zapytań oraz wybór baz danych (PubMed, Embase, Cochrane itp.)
- Krytyczna ocena dowodów klinicznych, wyników i dowodów naukowych.
- Podsumowanie dowodów na potrzeby dokumentacji regulacyjnej.
- Tworzenie dokumentów CER, CEP, PER i PEP
- Ocena luk w istniejących danych dokumentacji dotyczących CER/CEP/PEP/PER.
- Wykorzystanie zaawansowanych technik wyszukiwania w celu zebrania odpowiedniej literatury światowej

- Gwarantowana zgodność z rozporządzeniami MDR i IVDR
- Ustrukturyzowany, powtarzalny i uzasadniony proces wyszukiwania literatury
- Indywidualne strategie gromadzenia dowodów dostosowane do konkretnych urządzeń
- Wysoko wykwalifikowani eksperci w dziedzinie klinicznej i regulacyjnej
- Elastyczna wielkość zespołu; dostosowane do konkretnych urządzeń strategie oparte na dowodach naukowych
- Wkład różnych działów: regulacyjnego, medycznego i klinicznego
- End-to-end wsparcie w zakresie wyszukiwania End-to-end , przeglądu i dokumentacji
- Zwiększa wiarygodność, przejrzystość i gotowość dokumentacji przedkładanej organom regulacyjnym

Często Zadawane Pytania (FAQ)
01. Jaki jest cel protokołu wyszukiwania literatury dotyczącej wyrobów medycznych zgodnie z rozporządzeniami EU MDR?
Protokół wyszukiwania literatury dotyczącej wyrobów medycznych zapewnia uporządkowane, systematyczne i przejrzyste podejście do identyfikacji, oceny i dokumentowania dowodów naukowych dotyczących wyrobu lub produktów porównawczych. Gwarantuje on powtarzalność wyników, minimalizuje ryzyko stronniczości oraz umożliwia organom regulacyjnym prześledzenie sposobu gromadzenia, oceny i syntezy dowodów klinicznych lub dotyczących działania wyrobu. Zgodnie z EU MDR taki protokół wspiera ocenę bezpieczeństwa, działania oraz stosunku korzyści do ryzyka, zapewnia zgodność z przepisami i stanowi podstawę dla wysokiej jakości, uzasadnionej dokumentacji w całym cyklu życia wyrobu.
02. W jaki sposób najnowsze osiągnięcia naukowe wpływają na ocenę kliniczną i ocenę wyników?
Stan wiedzy na dzień dzisiejszy odzwierciedla aktualny poziom wiedzy naukowej i klinicznej dotyczącej danego typu wyrobu. Wyznacza on punkt odniesienia dla oczekiwanego poziomu bezpieczeństwa, działania i wyników klinicznych. Określenie stanu wiedzy na dzień dzisiejszy poprzez przegląd literatury pomaga umieścić twierdzenia dotyczące wyrobu w odpowiednim kontekście, ułatwia wybór produktów porównawczych oraz stanowi punkt odniesienia przy ocenie stosunku korzyści do ryzyka, planowaniu PMCF oraz aktualizacji danych dowodowych w trakcie cyklu życia wyrobu.
03. Czym różni się przegląd literatury w ramach MDR od tradycyjnego przeglądu systematycznego?
Przegląd literatury w ramach MDR/IVDR różni się od tradycyjnego przeglądu systematycznego tym, że koncentruje się na aspektach regulacyjnych i został opracowany specjalnie w celu zapewnienia zgodności z wymogami regulacyjnymi. Podczas gdy tradycyjne przeglądy systematyczne mają na celu udzielenie odpowiedzi na pytania badawcze i służą wyłącznie celom akademickim, przegląd literatury MDR ocenia dowody kliniczne i dotyczące działania w celu wykazania bezpieczeństwa wyrobu, jego działania oraz profilu korzyści i ryzyka. Opiera się on na ustrukturyzowanej, identyfikowalnej metodologii z z góry określonymi pytaniami badawczymi, kryteriami włączenia/wyłączenia oraz krytyczną oceną, aby stworzyć uzasadnioną, gotową do audytu dokumentację na potrzeby wniosków regulacyjnych.
04. Jak często należy aktualizować przeglądy literatury dotyczące wyroby medyczne wyrobów do diagnostyki in vitro?
Częstotliwość aktualizacji zależy od stopnia ryzyka związanego z wyrobem, sytuacji rynkowej oraz nowych danych naukowych. Wyroby wysokiego ryzyka zazwyczaj wymagają corocznych aktualizacji, podczas gdy w przypadku innych wyrobów można stosować ustalone odstępy czasowe. Przeglądy należy również aktualizować w przypadku pojawienia się istotnych sygnałów dotyczących bezpieczeństwa, nowych danych klinicznych, postępu technologicznego lub zmian w wytycznych, aby zapewnić dokładność profilu korzyści i ryzyka.
05. Jaką rolę odgrywają kryteria włączenia i wyłączenia w wyszukiwaniu literatury dotyczącej rozporządzeń MDR i IVDR?
Kryteria włączenia i wyłączenia gwarantują, że wybierane są wyłącznie istotne dane o wysokiej jakości. Zwiększają one obiektywność, ograniczają stronniczość recenzentów i zapewniają spójność procesu decyzyjnego. Zgodnie z rozporządzeniami MDR i IVDR kryteria te muszą być z góry określone, uzasadnione i dostosowane do pytań badawczych, aby zapewnić identyfikowalność i zgodność z przepisami w całym procesie oceny.
06. Dlaczego krytyczna ocena ma zasadnicze znaczenie w przeglądach literatury dotyczących rozporządzeń MDR i IVDR?
W ramach krytycznej oceny analizuje się jakość metodologiczną, trafność i wiarygodność uwzględnionych dowodów. Ramy regulacyjne MDR/IVDR kładą nacisk na tę ocenę, ponieważ organy regulacyjne opierają się na dobrze uzasadnionych oświadczeniach dotyczących bezpieczeństwa i działania wyrobów. Rygorystyczna ocena pomaga odróżnić solidne dane od wyników słabszych badań oraz wzmacnia wnioski wykorzystywane w analizach porównawczych (CER), ocenach porównawczych (PER), raportach z monitorowania po wprowadzeniu do obrotu (PMS) oraz analizach stosunku korzyści do ryzyka.
07. Czym różnią się wymogi dotyczące wyszukiwania literatury w ramach rozporządzeń MDR i IVDR?
Rozporządzenie MDR koncentruje się na ocenie klinicznej, uzasadnieniu stosunku korzyści do ryzyka oraz skuteczności klinicznej, podczas gdy rozporządzenie IVDR kładzie nacisk na skuteczność analityczną, zasadność naukową oraz skuteczność kliniczną w kontekście dokładności diagnostycznej. Strategie opracowywania literatury muszą odzwierciedlać te różnice poprzez dostosowanie pytań badawczych, zbiorów danych i ram oceny do odrębnych ścieżek dowodowych wymaganych w ramach każdego z tych rozporządzeń.
08. Jakie bazy danych i źródła informacji należy uwzględnić podczas wyszukiwania literatury zgodnego z wymogami MDR/IVDR?
Organy regulacyjne oczekują korzystania z wielu baz danych naukowych, takich jak PubMed, Embase i Cochrane, uzupełnionych o bazy danych dotyczące nadzoru, rejestry badań klinicznych, wytyczne oraz odpowiednią literaturę szarą. Korzystanie z różnorodnych źródeł zapewnia kompleksowy dostęp do informacji klinicznych, dotyczących działania i bezpieczeństwa, niezbędnych do rzetelnej oceny i bieżącego nadzoru.
09. Dlaczego firma Freyr jest uznawana za wiodącego partnera w zakresie wyszukiwania literatury i opracowywania protokołów?
Firma Freyr jest postrzegana jako wiodący partner dzięki dogłębnej znajomości przepisów, rygorystycznemu podejściu naukowemu oraz konsekwentnemu przestrzeganiu wymogów dotyczących dowodów określonych w rozporządzeniach MDR i IVDR. Zespół stosuje zasady systematycznego przeglądu, przejrzystą metodologię oraz wiedzę specjalistyczną w zakresie poszczególnych obszarów terapeutycznych, aby tworzyć uzasadnione i gotowe do kontroli wyniki. Podejście firmy Freyr kładzie nacisk na identyfikowalność, krytyczną ocenę oraz porównania z najnowszym stanem wiedzy – kluczowe czynniki cenione przez jednostki notyfikowane i organy regulacyjne na całym świecie.







