
USFDA Przegląd zatwierdzania wyrobów medycznych przed wprowadzeniem na rynek
Proces Zezwolenia przed wprowadzeniem do obrotu (PMA) USFDA to jedna ze ścieżek rejestracji wyrobów zapewnianych przez US FDA, przeznaczona głównie dla wyrobów medycznych klasy III FDA. Proces zatwierdzania PMA FDA dla wyrobów klasy III wymaga skrupulatnych ocen naukowych i regulacyjnych w celu oceny bezpieczeństwa i skuteczności wyrobu medycznego, zapewniając spełnienie najwyższych standardów przed dopuszczeniem do obrotu.
Umów spotkanie z naszymi ekspertami ds. zatwierdzeń przed wprowadzeniem na rynek
Kto powinien złożyć wniosek USFDA o zatwierdzenie wyrobu medycznego przed wprowadzeniem do obrotu (PMA)?
Producenci wyrobów muszą złożyć wniosek PMA, jeśli wyrób:
- Jest nowatorski.
- Należy do klasy wysokiego ryzyka.
- Nie można znaleźć w Bazie Danych Klasyfikacji Produktów.
- Nie jest zasadniczo równoważny (NSE) z wyrobami klasy I, II lub III.
Uzyskaj poradę eksperta w sprawie wniosku o zatwierdzenie przed wprowadzeniem do obrotu.
Jaka jest różnica między wnioskami 510(k), PMA i De-Novo?
Zezwolenie przed wprowadzeniem do obrotu
- Urządzenie należące do klasy III, które podtrzymuje życie ludzkie lub które stwarza potencjalne, nieuzasadnione ryzyko choroby lub obrażeń.
- Proces zatwierdzania PMA przez FDA wymaga badań klinicznych.
- Wymaga inspekcji na miejscu przed wydaniem zatwierdzenia PMA.
- 180 dni kalendarzowych
Klasyfikacja De Novo
- Nowatorskie urządzenia klasy I i II, które nie posiadają ważnego urządzenia referencyjnego.
- Wymaga danych z badań klinicznych.
- Brak audytu na miejscu przed zatwierdzeniem De-Novo.
- 150 dni kalendarzowych.
Rejestracja 510(k)
- Wyroby klasy III FDA, które wykazują znaczną równoważność z wyrobem referencyjnym.
- Nie wymaga testów na ludziach.
- Brak audytu na miejscu przed zatwierdzeniem 510(k).
- 90 dni kalendarzowych.
Jakie są różne metody składania wniosków o przedrynkowe zatwierdzenie FDA?
Producenci mogą wybrać jedną z poniższych czterech (04) metod składania wniosków PMA, która będzie najlepiej dopasowana do ich wyrobu:
- Tradycyjne PMA
- Modułowe PMA
- Protokół rozwoju produktu
- Wyłączenie dla Urządzeń Humanitarnych
Jakie są wymagania dotyczące danych do przedrynkowego zatwierdzenia wyrobów medycznych?
Zgodnie z 21 CFR część 814, wnioskodawcy muszą złożyć należycie wypełniony formularz wniosku CDRH, wymagane zobowiązania oraz dobrze przygotowany plik techniczny PMA do US FDA. Plik techniczny powinien zawierać dane niekliniczne i kliniczne.
Dane niekliniczne – Obejmują one dane dotyczące mikrobiologii, toksykologii, immunologii, biokompatybilności, wytrzymałości, zużycia, okresu przydatności do użycia oraz innych badań laboratoryjnych lub na zwierzętach.
Dane kliniczne – Obejmują one dane dotyczące protokołów badań, dane dotyczące bezpieczeństwa i skuteczności, działań niepożądanych i powikłań, awarii i wymian urządzeń, informacji o pacjentach, skarg pacjentów, zestawienia danych od wszystkich poszczególnych badanych, wyników analiz statystycznych oraz wszelkich innych informacji z badań klinicznych.
Czym jest Proces składania wniosku PMA?
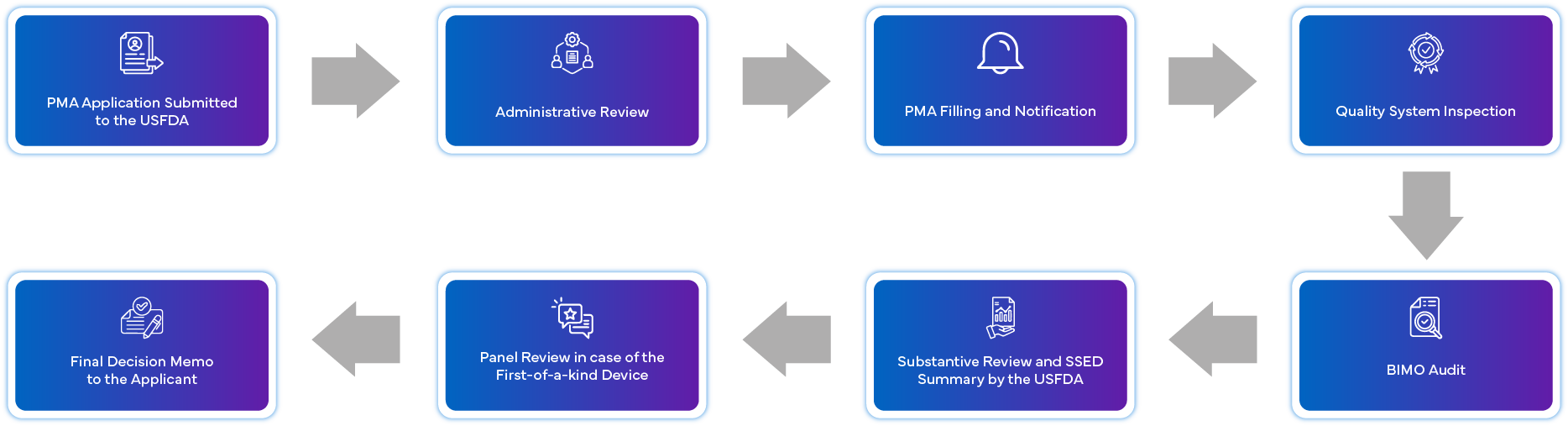
Jakie są wymogi zgodności po zatwierdzeniu dla PMA?
Urządzenia zatwierdzone w ramach ścieżki PMA muszą być zgodne z wymaganiami po wprowadzeniu do obrotu określonymi przez USFDA. Urządzenie musi być zgodne z następującymi:
- Wymagania po zatwierdzeniu nałożone w decyzji zatwierdzającej FDA PMA.
- Zarządzanie zmianami po zatwierdzeniu poprzez terminowe złożenie odpowiednich suplementów PMA
- Zgłoszenie raportów po zatwierdzeniu (rocznych).
- Przepisy 21 CFR 803 dotyczące Raportowania Wyrobów Medycznych (MDR)
- Badania nadzoru po wprowadzeniu do obrotu zgodnie z wymogami USFDA w decyzjach o zatwierdzeniu PMA.
Jakie są opłaty USFDA za rozpatrzenie wniosku PMA?
Opłaty użytkownika MDUFA za oryginalne PMA i suplementy są następujące:
| Rodzaj wniosku | Opłaty za rok fiskalny 2023 (od 1 października 2022 do 30 września 2023) | |
|---|---|---|
| Standardowa opłata | Opłata dla małych przedsiębiorstw | |
| PMA, PDP, PMR, BLA | $441,547 |
|
| Uzupełnienie w trybie Panel-Track | $353,238 | $88,309 |
| 180-dniowy Suplement | $66,232 | $16,558 |
| Roczna opłata za raportowanie okresowe dotyczące wyrobu klasy III (PMAs, PDPs i PMRs) | $15,454 | $3,864 |
| 30-dniowe zawiadomienie | $7,065 | $3,532 |
| Uzupełnienie w czasie rzeczywistym | $30,908 | $7,727 |
Dzięki doświadczeniu w obsłudze zgłoszeń PMA, Freyr może pomóc w identyfikacji i kompilacji informacji oraz w przygotowaniu i przeglądzie wniosku.
USFDA Ekspertyza i zalety w zakresie zatwierdzania wyrobów medycznych przed wprowadzeniem na rynek
- Należyta staranność regulacyjna
- Zgodność z inspekcją systemu jakości
- Zgodność z audytem BIMO
- Opracowanie dokumentacji technicznej PMA
- Publikowanie i tworzenie eCopy
- Walidacja i złożenie eCopy.
- Odpowiada na Odpowiedź RTA i niedociągnięcia
- Usługi pośrednictwa do momentu uzyskania zgody FDA na wprowadzenie do obrotu.
- Konsultacje w sprawie braków
- Wykaz urządzeń i rejestracja zakładu
- Zarządzanie uzupełnieniami PMA i 30-dniowymi powiadomieniami
- Coroczne składanie raportów okresowych
- Audyty próbne i szkolenia z zakresu 21 CFR 820

- Doświadczenie w obsłudze wielu zgłoszeń PMA do FDA dla zróżnicowanych kategorii wyrobów.
- Zespół ekspertów do wniosków o przedrynkowe zatwierdzenie FDA zgodnie z wymaganiami regulacyjnymi.
- Dodatkowe wsparcie w obsłudze zapytań związanych z PMA.
- Terminowe złożenie rezultatów
- Na bieżąco z nowymi poprawkami US FDA
