


Rejestracja wyrobów medycznych w Indonezji – przegląd
Indonezja wprowadziła powszechną opiekę zdrowotną dla swoich obywateli w 2014 roku. Znacząco wpłynęło to na rozwój rynku wyrobów medycznych i doprowadziło do wzrostu importu wyrobów medycznych. Wyroby medyczne w Indonezji są regulowane przez Narodową Agencję Kontroli Leków i Żywności (NADFC), działającą pod nadzorem indonezyjskiego Ministerstwa Zdrowia (MoH). Najnowszym obowiązującym rozporządzeniem dotyczącym importu wyrobów medycznych jest Dekret nr 62 wprowadzony w 2017 roku. Firmy zagraniczne muszą wyznaczyć lokalnego autoryzowanego przedstawiciela w Indonezji do procesu rejestracji wyrobów medycznych w Indonezji.
![]()
Organ regulacyjny: Krajowa Agencja Kontroli Leków i Żywności (NADFC)![]()
Regulacja: Nr 62 / 2017![]()
Autoryzowany Przedstawiciel: Lokalny upoważniony przedstawiciel w Indonezji![]()
Wymóg QMS: ISO 13485:2016![]()
Ocena Danych Technicznych: NADFC![]()
Ważność licencji: 5 Lat![]()
Wymogi dotyczące oznakowania: Nr 62 / 2017![]()
Format przedłożenia: Online/Papierowo![]()
Język: Angielski i indonezyjski
Klasyfikacja wyrobów medycznych w Indonezji
Obecne przepisy klasyfikują urządzenia jako A, B, C i D na podstawie ryzyka.
| Kryteria Ryzyka | Klasa wyrobu |
|---|---|
| Niskie ryzyko | A |
| Niskie do umiarkowanego ryzyko | B |
| Ryzyko umiarkowane – wysokie | C |
| Wysokie ryzyko | D |
Lokalny upoważniony przedstawiciel w Indonezji
Indonezyjskie przepisy wymagają od producentów wyznaczenia lokalnego przedstawiciela posiadającego licencję dystrybutora. Dystrybutor może zostać wyznaczony do reprezentowania zagranicznego producenta w Indonezji. Jednak wyznaczenie niezależnej strony trzeciej zapewniłoby elastyczność w zmianie dystrybutorów lub wyznaczeniu wielu dystrybutorów w celu lepszej penetracji rynku.
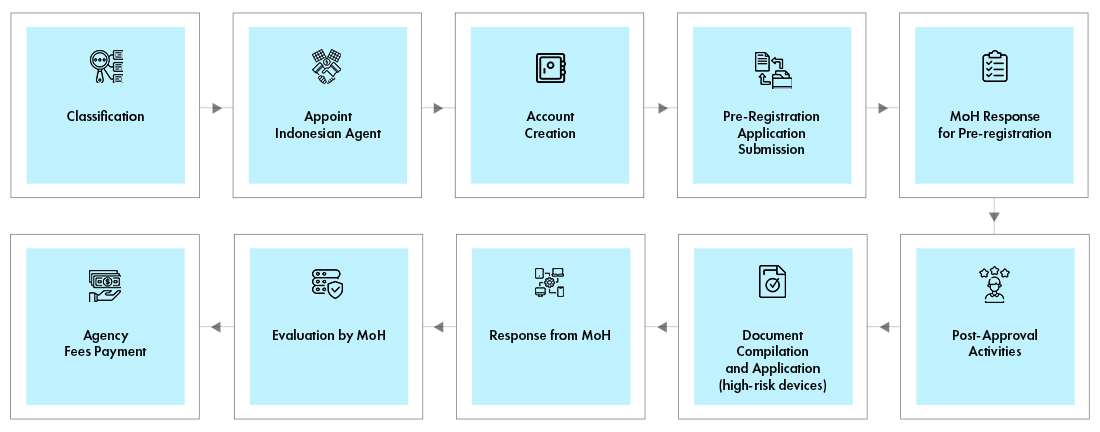
Rejestracja wyrobów medycznych w Indonezji
Lokalny przedstawiciel musi założyć konto na portalu internetowym. Proces rejestracji jest taki sam dla wszystkich klas urządzeń. Jednakże wymagania dotyczące dokumentacji różnią się w zależności od klasy urządzenia. Rejestracja jest procesem dwuetapowym –
- Proces przedrejestracyjny
- Proces oceny
MoH weryfikuje klasyfikację wyrobu i określa koszt oceny. Wynik wstępnej rejestracji wraz z fakturą jest przesyłany drogą elektroniczną do wnioskodawcy. Lokalny przedstawiciel, w imieniu producenta, dokonuje płatności i przesyła dowód wpłaty. MoH przegląda dokumenty i przekazuje wyniki wnioskodawcy drogą elektroniczną. Niektóre wyroby wymagają testów krajowych w akredytowanym laboratorium.
Przegląd procesu zatwierdzania regulacyjnego
Zespół ekspertów Freyr śledzi zmieniające się trendy i przepisy oraz pomaga zainteresowanym stronom w utrzymaniu zgodności regulacyjnej przez cały cykl życia produktu. Oferujemy rozwiązania regulacyjne, aby utrzymać inne aspekty zgodności regulacyjnej w ramach ograniczonych budżetów.
Klasa wyrobu | Klasa ryzyka | Terminy MoH dla Pozwolenie na dopuszczenie do obrotu produktu leczniczego. | Terminy MoH dla Odnowienie / Zmiana | ||
|---|---|---|---|---|---|
| Proces klasyfikacji (Dni) | Proces oceny (Dni) | Proces klasyfikacji (Dni) | Proces oceny (Dni) | ||
| Klasa A | Niskie ryzyko | 7 | 45 | 7 | 45 |
| Klasa B | Niskie do umiarkowanego ryzyko | 7 | 90 | 7 | 45 |
| Klasa C | Ryzyko umiarkowane – wysokie | 7 | 100 | 7 | 45 |
| Klasa D | Wysokie ryzyko | 7 | 120 | 7 | 45 |
Ekspertyza Freyr
- Należyta staranność regulacyjna
- Rejestracja wyrobów
- Badania w kraju
- Licencjonowanie dystrybutorów
- Legalizacja i notarialne poświadczenie
- Przedstawiciel prawny
- Wsparcie w zakresie oznakowania
- Wsparcie tłumaczeniowe
- Identyfikacja i kwalifikacja dystrybutora
- Usługi nadzoru po wprowadzeniu do obrotu
- Zarządzanie zmianami po zatwierdzeniu
- Usługi odnowienia i przeniesienia licencji
- Usługi składania wniosków i koordynacji
