Rejestracja wyrobów medycznych w ZEA – przegląd
Zjednoczone Emiraty Arabskie (ZEA), znaczący kraj członkowski GCC, posiadają zaawansowany system opieki zdrowotnej. Ich potencjał rynkowy jest udowodniony i stale rośnie, a nadzór nad nim sprawuje Departament Kontroli Leków Ministerstwa Zdrowia i Prewencji (MOHAP). Scentralizowane zarządzanie i bariery językowe stanowią główne przeszkody w rejestracji wyrobów medycznych w ZEA, podobnie jak złożoności językowe i brak efektywnych kanałów komunikacji z organami ds. zdrowia.
![]()
Organ regulacyjny: Departament Kontroli Leków Ministerstwa Zdrowia i Prewencji (MOHAP)![]()
Regulacja: Wytyczne dotyczące rejestracji wyrobów medycznych w ZEA![]()
Ścieżka regulacyjna: Rejestracja produktu![]()
Autoryzowany Przedstawiciel: Wymagany jest lokalny autoryzowany przedstawiciel w ZEA![]()
Wymóg QMS: ISO 13485:2016![]()
Ocena Danych Technicznych: Komitet ds. Rejestracji Wyrobów Medycznych![]()
Ważność licencji: 5 Lat![]()
Wymogi dotyczące oznakowania: Załącznik 2 (2.5) Wytycznych ZEA dotyczących rejestracji wyrobów medycznych![]()
Format przedłożenia: Papier![]()
Język: Angielski
Klasyfikacja wyrobów medycznych w ZEA
ZEA posiada odrębne zasady klasyfikacji dla wyrobów medycznych i wyrobów medycznych do diagnostyki in vitro (IVD). Zasady klasyfikacji wyrobów medycznych w ZEA są zgodne z zasadami klasyfikacji dyrektyw UE dotyczących wyrobów medycznych. Klasy wyrobów zgodnie z zasadami klasyfikacji ZEA są następujące
| Kryteria Ryzyka | Klasa wyrobu medycznego |
|---|---|
| Niskie ryzyko | I |
| Niskie do umiarkowanego ryzyko | IIa |
| Ryzyko umiarkowane – wysokie | IIb |
| Wysokie ryzyko | III |
| Kryteria Ryzyka | Klasa IVD |
|---|---|
| Niskie ryzyko indywidualne i niskie ryzyko dla zdrowia publicznego | A |
Umiarkowane ryzyko indywidualne i/lub Niskie Ryzyko dla Zdrowia Publicznego | B |
Wysokie ryzyko indywidualne i/lub Umiarkowane ryzyko dla zdrowia publicznego | C |
| Wysokie ryzyko indywidualne i wysokie ryzyko dla zdrowia publicznego | D |
Lokalny autoryzowany przedstawiciel w ZEA
Zagraniczni producenci, nieposiadający fizycznego biura, powinni wyznaczyć Lokalnego Przedstawiciela (LR) do działania w ich imieniu. Lokalny przedstawiciel powinien posiadać licencję Ministerstwa Zdrowia jako sklep medyczny lub biuro naukowe (w przypadku biura naukowego, działania importowe i dystrybucyjne powinny być wykonywane przez wyznaczony Licencjonowany Sklep Medyczny). Wnioskodawcy mogą wyznaczyć swojego dystrybutora jako Lokalnego Przedstawiciela. Jednak posiadanie niezależnego Lokalnego Przedstawiciela, bez interesów handlowych, zapewniłoby wymaganą elastyczność w wyznaczaniu wielu dystrybutorów w ZEA. Szczegóły zarówno LR, jak i dystrybutora muszą być podane podczas rejestracji wyrobu.
Oficjalny proces klasyfikacji z UAE MoHAP
Ministerstwo Zdrowia i Prewencji ZEA (UAE MoHAP) wprowadziło oficjalną usługę klasyfikacji, szczególnie przydatną, gdy nie masz pewności, czy Twój produkt wymaga rejestracji. Usługa ta klasyfikuje produkty wszystkich typów i form na podstawie ich prezentacji, składu, zastosowania i projektu. Wymagania mogą się różnić w zależności od charakteru produktu, klasy ryzyka i statusu regulacyjnego.
Pismo klasyfikacyjne wskazuje, czy produkt musi być zarejestrowany w MoHAP. Jeśli rejestracja jest wymagana, produkt musi zostać zarejestrowany zgodnie z klasą określoną w piśmie klasyfikacyjnym. Pismo to jest ważne przez trzy lata od daty wydania.
Oficjalne wyniki klasyfikacji mogą być następujące:
- Nie wymaga rejestracji MOHAP
- Zatwierdzony przez MOHAP ZEA jako wyrób medyczny, przeznaczony wyłącznie do użytku profesjonalnego
- Zatwierdzony przez MOHAP ZEA jako wyrób medyczny dostępny bez recepty
Rejestracja wyrobów medycznych w ZEA
Niektóre wyroby nie wymagają rejestracji produktu ani wcześniejszego wpisu lub zatwierdzenia do importu. Produkty zwolnione z rejestracji lub wpisu muszą złożyć wniosek i uzyskać pozwolenie na import, aby mogły być wprowadzane do obrotu w ZEA.
W przypadku innych wyrobów, import nie zostanie dopuszczony, chyba że DRCD wyda wstępną zgodę na import danej partii. Takie wyroby muszą być wymienione lub zarejestrowane, aby mogły być importowane do ZEA.
Wpis do wykazu urządzeń: Zazwyczaj produkty używane w szpitalach pod nadzorem specjalistów oraz urządzenia klasy I nie podlegają szczegółowej ocenie i wymagają wpisu do wykazu. Agencja wyda Certyfikat Wpisu do Wykazu. Urządzenia po wpisaniu do wykazu muszą uzyskać pozwolenie na import, aby móc wprowadzać je na rynek w ZEA.
Rejestracja wyrobów: Działalność rejestracyjna obejmuje rejestrację zakładu i produktu.
- Rejestracja zakładu: Zakład produkcyjny musi być zarejestrowany, jeśli wyrób wyprodukowany w tym zakładzie jest po raz pierwszy importowany do ZEA. W przypadku kolejnych wyrobów wyprodukowanych w tym samym zakładzie wystarczy jedynie rejestracja wyrobu, a rejestracja zakładu nie jest wymagana.
- Rejestracja urządzeń: Urządzenia te podlegają ocenie przez komisję techniczną, która po zatwierdzeniu wyda świadectwo licencyjne.
Przebieg procesu
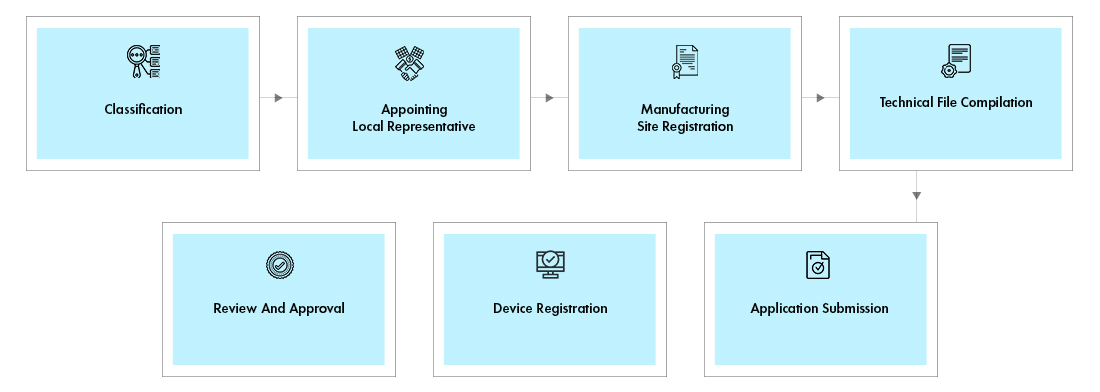
Zarządzanie cyklem życia urządzenia po zatwierdzeniu
- Zarządzanie zmianami po zatwierdzeniu – modyfikacje istniejących zatwierdzeń wyrobów medycznych, takie jak dodawanie nowych wariantów, akcesoriów; dodawanie nowych wskazań do stosowania i inne
- Utrzymywanie zatwierdzeń i rejestracji poprzez terminowe uiszczanie opłat administracyjnych i rejestracyjnych
- Odnowienie licencji
- Pośrednictwo między MoH a producentem
- Zarządzanie importem
Dzięki wyłącznemu centrum dostaw w Dubaju, Freyr ma dominującą pozycję na rynku wyrobów medycznych w ZEA oraz określa klasyfikację wyrobów medycznych, a także interpretuje przepisy wytycznych w celu lepszego zapewnienia zgodności. Wspieramy klientów w kompilacji dokumentacji zgodnie ze standardami, co zapewnia szybkie zatwierdzenia. Freyr oferuje pełen zakres usług regulacyjnych związanych ze skutecznym wprowadzaniem wyrobów medycznych na rynek.
Podsumowanie
| Rodzaj urządzenia | Wykaz urządzeń | Rejestracja wyrobów | Licencja Importowa |
|---|---|---|---|
Wyrób zwolniony z zatwierdzenia przedimportowego (Zgodnie z wykazem w Załączniku 3) | N/D | N/D | Tak |
| Tak | N/D | Tak |
| Wszystkie pozostałe urządzenia | N/D | Tak | Tak |
Ekspertyza Freyr
- Wywiad regulacyjny
- Należyta staranność regulacyjna
- Formalna klasyfikacja wyrobów medycznych
- Rejestracja wyrobów
- Autoryzowana reprezentacja w ZEA
- Wsparcie tłumaczeniowe
- Wsparcie w zakresie etykietowania
- Identyfikacja i kwalifikacja dystrybutora
- Zarządzanie zmianami po zatwierdzeniu
- Odnowienie i przeniesienie licencji
- Odprawa celna