3 minuty czytania
BIMO oznacza Bioresearch Monitoring (Monitorowanie Badań Biologicznych), program inspekcji na miejscu i audytów danych, mający na celu monitorowanie wszystkich aspektów prowadzenia i raportowania badań regulowanych przez Amerykańską Agencję ds. Żywności i Leków (FDA). Program został ustanowiony w 1977 roku po zidentyfikowaniu potrzeby audytowania miejsc badań klinicznych. Głównym celem tego programu jest zapewnienie jakości i integralności danych przedstawianych do zatwierdzeń nowych produktów i wniosków marketingowych. Ponadto program ten chroni prawa i dobro zarówno ludzi, jak i zwierząt uczestniczących w badaniach regulowanych przez FDA.
Główne cele Programu BIMO
Rocznie przeprowadza się ponad 1000 inspekcji. Główne cele realizowane w ramach programu BIMO to:
- Audyt danych klinicznych
- Inspekcja trwających badań klinicznych
- Inspekcja laboratoriów nieklinicznych
- Inspekcja Komisji Bioetycznych (IRB)
Które produkty wchodzą w zakres audytu BIMO?
BIMO ma zastosowanie do leków, produktów biologicznych, wyrobów medycznych, produktów spożywczych, wyrobów tytoniowych i produktów weterynaryjnych. Program zgodności jest nadzorowany przez sześć (06) centrów produktowych FDA – Centrum Oceny i Badań Biologicznych (CBER), Centrum Wyrobów Medycznych i Zdrowia Radiologicznego (CDRH), Centrum Oceny i Badań Leków (CDER), Centrum Bezpieczeństwa Żywności i Stosowanego Żywienia (CFSAN), Centrum Produktów Tytoniowych (CTP) oraz Centrum Medycyny Weterynaryjnej (CVM).
Które firmy podlegają audytowi BIMO?
Zarówno firmy krajowe, jak i międzynarodowe, prowadzące którąkolwiek z poniższych działalności lub podlegające im, podlegają wymogom monitorowania badań biologicznych –
- Laboratoria badań nieklinicznych dla zgodności z Dobrą Praktyką Laboratoryjną (GLP)
- Badacze kliniczni w celu zapewnienia zgodności z Dobrą Praktyką Kliniczną (GCP)
- Sponsorzy
- Organizacje Badawczo-Kontraktowe (CRO)
- Monitorzy badań klinicznych
- Obiekty do badań biorównoważności in vivo
- Komisje Bioetyczne (IRBs)
Które programy zgodności wchodzą w zakres Programu BIMO?
US FDA może przeprowadzić audyt BIMO w dowolnym momencie za pośrednictwem siedmiu (07) wieloośrodkowych programów zgodności. Te siedem wieloośrodkowych programów zgodności jest wdrażanych poprzez –
- Inspekcja Badacza Klinicznego (CI) i Badacza Sponsora (SI)
- Inspekcja Komisji Bioetycznej (IRB)
- Inspekcja Organizacji Badań Kontraktowych/Sponsora/Monitora (CRO/S/M)
- Inspekcja Dobrej Praktyki Laboratoryjnej (GLP)
- Inspekcja Biorównoważności-Biodostępności (BEQ)
- Inspekcja dotycząca zgłaszania niepożądanych zdarzeń po wprowadzeniu leku na rynek (PADE).
- Inspekcja Raportowania Strategii Oceny i Łagodzenia Ryzyka (REMS)
Każdy z tych programów określa szczegółowy zakres przeglądu lub inspekcji, które należy przeprowadzić, aby zapewnić zgodność z FDA.
Jakie przepisy mają zastosowanie do audytu BIMO?
Przepisy – 21 CFR 50 – Ochrona Podmiotów Ludzkich, 21 CFR 54 – Ujawnianie Informacji Finansowych, 21 CFR 56 – IRBs, 21 CFR 58 – Dobra Praktyka Laboratoryjna dla laboratoriów nieklinicznych, 21 CFR 809 – Produkty do Diagnostyki In Vitro oraz 21 CFR 812 – Wyłączenie dla Badanych Wyrobów Medycznych mają zastosowanie do audytu BIMO.
Ile audytów jest przeprowadzanych rocznie w ramach Programu BIMO?
Liczba audytów BIMO przeprowadzanych przez US FDA zmienia się każdego roku. W ostatnich latach liczba kontroli na miejscu spadła z powodu pandemii COVID-19, a FDA musiała wstrzymać cały nadzór na miejscu nad badaniami klinicznymi. Monitorowano jedynie konkretne, krytyczne i kluczowe badania kliniczne.
„Zdalne Oceny Regulacyjne” (RRA) zostały wprowadzone podczas pandemii COVID-19 w celu zdalnego monitorowania regulowanych badań. RRA są przeprowadzane za pośrednictwem wideokonferencji i są dobrowolną inicjatywą do zdalnej oceny danych i procesów. Należy jednak zauważyć, że RRA nie są równoważne ani alternatywne dla inspekcji na miejscu, lecz są jedynie procedurą, która ewoluowała z powodu pandemii COVID-19.
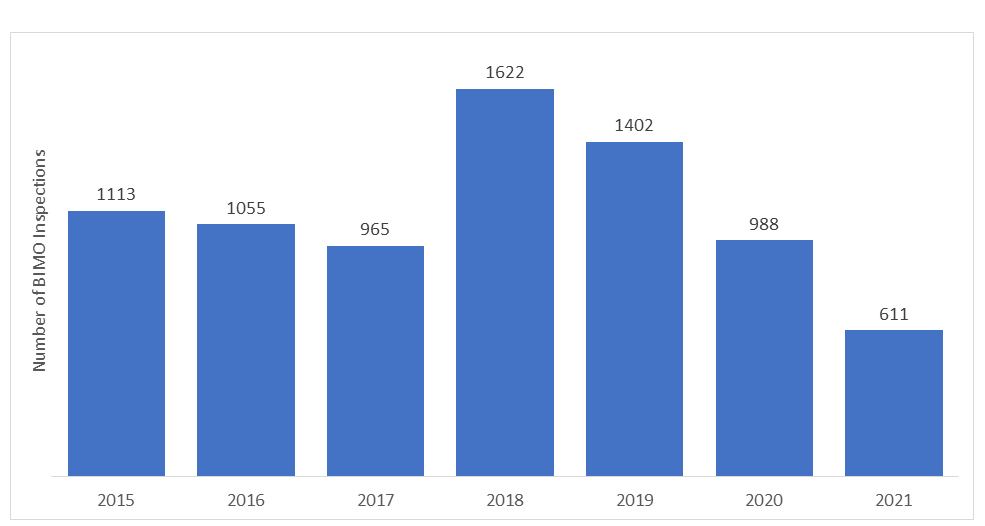
*Dane przedstawione za lata 2020 i 2021 nie obejmują inspekcji RRA
Ile zdalnych ocen regulacyjnych (RRAs) przeprowadzono podczas pandemii COVID-19 w ramach Programu BIMO?
W 2021 roku znacząco wzrosło zastosowanie inspekcji RRA we wszystkich programach. W kwietniu 2021 roku FDA opublikowała dokument wytycznych pt. „Zdalne interaktywne oceny zakładów produkujących leki i monitorujących badania biologiczne podczas stanu zagrożenia zdrowia publicznego COVID-19 – Wytyczne dla przemysłu”, który zawiera wyczerpujące informacje na temat procesu przeprowadzania RRA przez FDA.
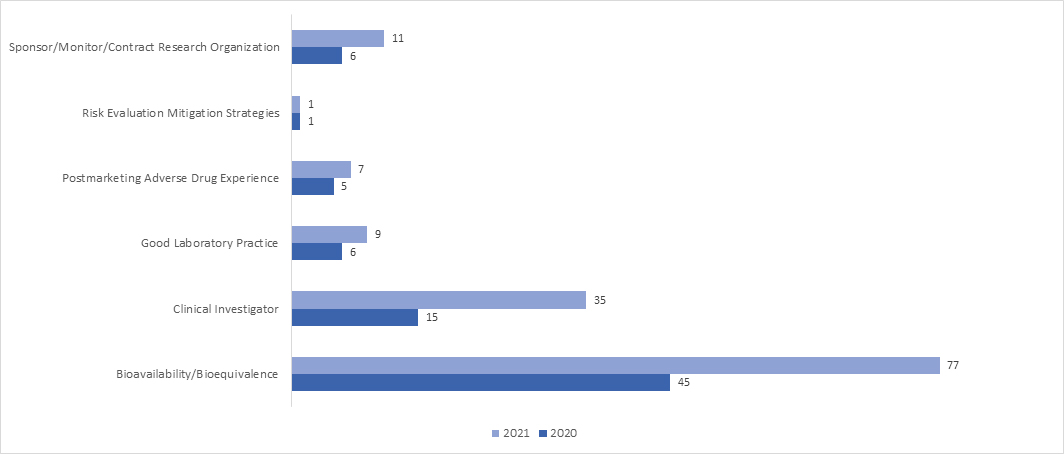
Jakie są możliwe wyniki audytu BIMO?
Podczas audytu BIMO, US FDA może podjąć jedną z poniższych działań w zależności od zgodności –
1. Nie wskazano żadnych działań (NAI)
NAI ma zastosowanie, gdy inspektor terenowy FDA nie stwierdził żadnych nieprawidłowości lub jedynie drobne problemy, dla których dalsze działania nie są uzasadnione.
2. Wskazane Działanie Dobrowolne (VAI)
VAI ma zastosowanie, gdy zidentyfikowano niepożądane praktyki, ale nie są one znaczące.
3. Wskazane Działanie Urzędowe (OAI)
OAI ma zastosowanie, gdy zidentyfikowano nieakceptowalne praktyki, które naruszają integralność danych i/lub prawa osób badanych.
Jakie są najczęstsze niezgodności stwierdzone podczas audytu BIMO?
Niektóre z najczęstszych niezgodności zaobserwowanych podczas audytu BIMO to –
- Brak właściwego prowadzenia ewidencji
- Niezgodność z planem badawczym
- Niezgodność z przepisami
- Niewłaściwe monitorowanie protokołów
- Niewystarczająca ochrona uczestników badania
- Niewystarczająca odpowiedzialność za badany produkt
Audyt BIMO jest kluczowy dla każdego dewelopera lub producenta nowych wyrobów medycznych i technologii planującego wprowadzenie swojego wyrobu na rynek US. Przestrzeganie przepisów i wytycznych, aby uniknąć opisanych pułapek, jest bardzo ważne.
Potrzebujesz pomocy w zakresie inspekcji audytowych BIMO? Skontaktuj się z Freyr. Bądź na bieżąco. Zachowaj zgodność.