
Protocolo de búsqueda Dispositivos Médicos y resumen de la revisión sobre diagnósticos in vitro (IVD) y Dispositivos Médicos
En el complejo panorama de los productos sanitarios y los productos para diagnóstico in vitro (IVD), un protocolo de búsqueda bibliográfica bien estructurado para los productos sanitarios es más que un simple ejercicio de investigación: es un requisito fundamental para cumplir con EU MDR y el Reglamento (UE) 2017/746 sobre productos para diagnóstico in vitro.
Una revisión exhaustiva de la literatura médica respalda las evaluaciones clínicas y de rendimiento, las actividades posteriores a la comercialización y las solicitudes reglamentarias. Permite la transparencia, la trazabilidad y la reproducibilidad, aspectos esenciales tanto para cumplir con las expectativas reglamentarias como para garantizar la localizabilidad de la evidencia generada por la IA, elementos clave destacados por los organismos reguladores internacionales y fundamentales para la localizabilidad de las búsquedas basadas en la IA.
Requisitos de «estado de la técnica» según EU MDR el IVDR EU MDR
Tanto en EU MDR en el Reglamento sobre productos de diagnóstico in vitro (IVDR) de EU MDR , la determinación del estado actual de la técnica (SOTA) es un requisito obligatorio para la evaluación clínica y de rendimiento. El SOTA representa el nivel actual y generalmente aceptado de conocimientos científicos, técnicos y clínicos relevantes para el producto sanitario o el producto de diagnóstico in vitro.
Es fundamental contar con un protocolo de búsqueda bibliográfica sólido para:
![]() Identificar las tecnologías de referencia y los estándares terapéuticos
Identificar las tecnologías de referencia y los estándares terapéuticos![]() Establecer perfiles de seguridad y rendimiento aceptados
Establecer perfiles de seguridad y rendimiento aceptados![]() Compara el dispositivo en cuestión con las alternativas actuales
Compara el dispositivo en cuestión con las alternativas actuales![]() Apoyar las actividades de CER, PER, CEP, PEP, PMS y PMCF
Apoyar las actividades de CER, PER, CEP, PEP, PMS y PMCF![]() Recopilar pruebas sólidas que justifiquen la relación beneficio-riesgo
Recopilar pruebas sólidas que justifiquen la relación beneficio-riesgo
 Identificar las tecnologías de referencia y los estándares terapéuticos
Identificar las tecnologías de referencia y los estándares terapéuticos Establecer perfiles de seguridad y rendimiento aceptados
Establecer perfiles de seguridad y rendimiento aceptados Compara el dispositivo en cuestión con las alternativas actuales
Compara el dispositivo en cuestión con las alternativas actuales Apoyar las actividades de CER, PER, CEP, PEP, PMS y PMCF
Apoyar las actividades de CER, PER, CEP, PEP, PMS y PMCF Recopilar pruebas sólidas que justifiquen la relación beneficio-riesgo
Recopilar pruebas sólidas que justifiquen la relación beneficio-riesgoFreyr garantiza que su dossier de documentación demuestre plenamente el cumplimiento de las expectativas de SOTA, un factor clave para la aceptación por parte del organismo notificado.
Revisión EU MDR sobre el Reglamento de la UE relativo a los productos de diagnóstico in vitro (IVDR) y el Reglamento EU MDR )
La revisión de la literatura del EU IVDR/ EU MDR es un componente crítico en la gestión del ciclo de vida de un Dispositivo Médico o IVD. Una estrategia sistemática de búsqueda de literatura del EU IVDR/ EU MDR proporciona la base para los Clinical evaluation reports (CER), los Informes de Evaluación del Rendimiento (PER), la Vigilancia Post-Comercialización (PMS), las actividades de PMCF/PMPF ancladas en la literatura basada en evidencia de IVD / Dispositivos Médicos, este proceso permite a los fabricantes apoyar la evaluación continua de la seguridad y el rendimiento.
La revisión EU MDR sobre el IVDR y EU MDR suele incluir:
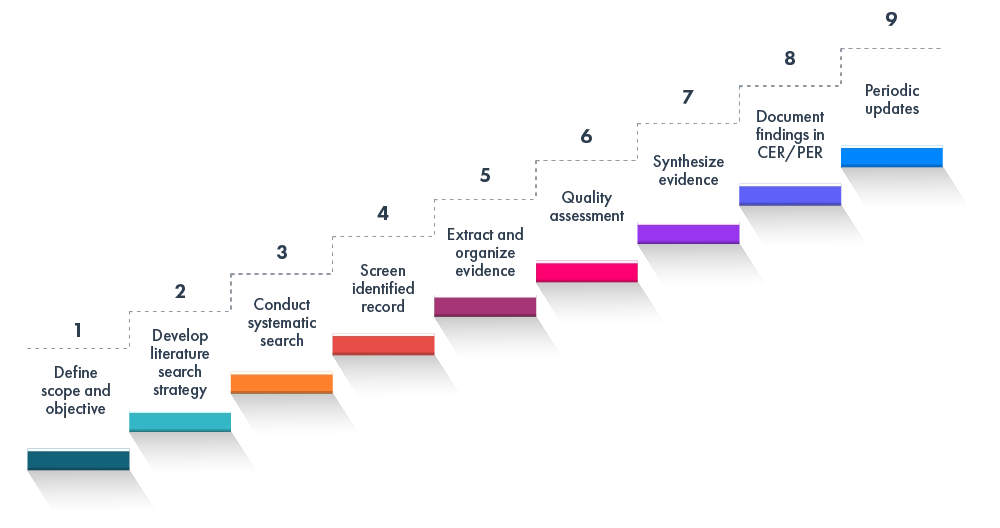
Este marco se ajusta a las mejores prácticas internacionales en materia de evaluaciones clínicas y de rendimiento, así como de revisiones bibliográficas.
Diferencias clave entre los requisitos de búsqueda bibliográfica del IVDR y del MDR
Aunque el MDR y el IVDR comparten una base común de evaluación sistemática de la evidencia, sus requisitos difieren
El Reglamento sobre productos sanitarios (MDR) se centra en
![]() Evaluación clínica y evidencia clínica
Evaluación clínica y evidencia clínica![]() Declaraciones sobre seguridad y rendimiento
Declaraciones sobre seguridad y rendimiento![]() Recopilación de PMCF
Recopilación de PMCF![]() Justificación de la relación beneficio-riesgo
Justificación de la relación beneficio-riesgo![]() Ajustes según MEDDEV 2.7/1 Rev. 4
Ajustes según MEDDEV 2.7/1 Rev. 4
 Justificación de la relación beneficio-riesgo
Justificación de la relación beneficio-riesgoEl Reglamento sobre productos sanitarios para diagnóstico in vitro (IVDR) se centra en
![]() Validez científica
Validez científica![]() Rendimiento analítico
Rendimiento analítico![]() Desempeño clínico
Desempeño clínico![]() Desarrollo de PER y PEP
Desarrollo de PER y PEP![]() Requisitos de pruebas del PMPF
Requisitos de pruebas del PMPF![]() Una reclasificación más estricta, que exige más pruebas justificativas
Una reclasificación más estricta, que exige más pruebas justificativas
 Validez científica
Validez científica Rendimiento analítico
Rendimiento analítico Desarrollo de PER y PEP
Desarrollo de PER y PEP Requisitos de pruebas del PMPF
Requisitos de pruebas del PMPF Una reclasificación más estricta, que exige más pruebas justificativas
Una reclasificación más estricta, que exige más pruebas justificativasFreyr adapta las estrategias de búsqueda bibliográfica, la elaboración de estudios de experiencia clínica (CER) y de experiencia postcomercialización (PER), así como los protocolos de seguimientoPMCF) y seguimiento de la seguridad y eficacia a largo plazo (PMPF), en función de la vía regulatoria del producto sanitario.
El poder de un equipo sólido de síntesis de literatura científica
Cumplir con los requisitos del MDR y el IVDR exige algo más que simples búsquedas en bases de datos. Un equipo especializado en la síntesis de literatura científica, con experiencia en el ámbito terapéutico, garantiza que su revisión bibliográfica del IVDR/MDR, su protocolo de búsqueda bibliográfica y la documentación de la evaluación clínica y de rendimiento cumplan con el nivel de profundidad y rigor que exigen las autoridades reguladoras.
Los expertos de Freyr simplifican los procesos complejos y transforman los datos clínicos, de rendimiento y científicos en pruebas claras y fundamentadas que refuerzan los protocolos de búsqueda bibliográfica, los CER, los PER y las estrategias de PMS/PMPF.
Gracias a metodologías sistemáticas, técnicas de búsqueda avanzadas y habilidades de evaluación crítica, nuestro equipo se asegura de que cada revisión bibliográfica cumpla con las expectativas normativas internacionales, al tiempo que mejora la calidad, la credibilidad y la solidez de su dossier de evidencia, lo que le proporciona a su dispositivo una sólida ventaja competitiva en un mercado en rápida evolución.
Protocolo de búsqueda EU MDR sobre el Reglamento de la UE sobre productos de diagnóstico in vitro (IVDR) y el Reglamento EU MDR )
Un protocolo de búsqueda EU MDR conforme al Reglamento sobre productos sanitarios para diagnóstico in vitro (IVDR) y al Reglamento sobre productos sanitarios EU MDR aporta estructura, reduce el sesgo de los revisores y garantiza una trazabilidad completa durante las auditorías.
El proceso del protocolo de búsqueda bibliográfica sobre el IVDR y el MDR incluye:
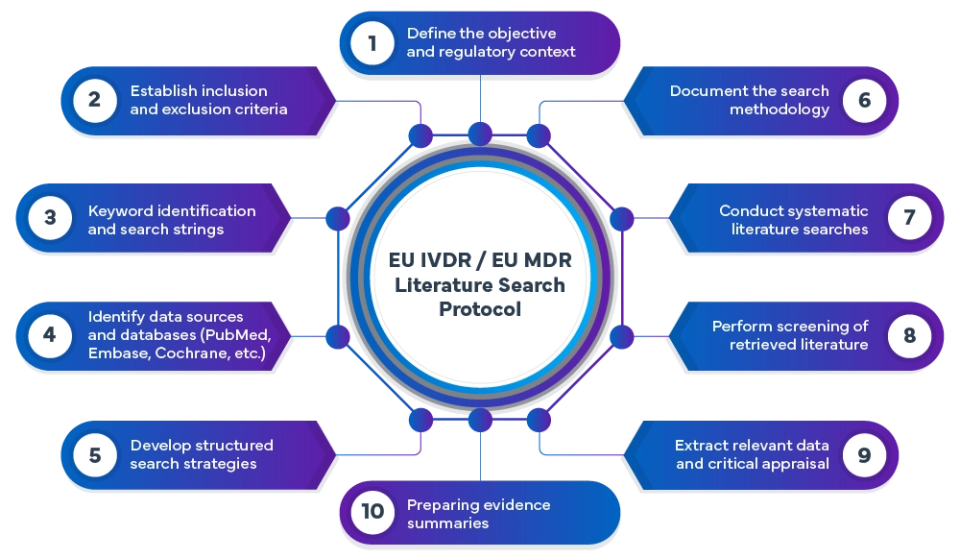
En Freyr, llevamos a cabo revisiones bibliográficas exhaustivas que cumplen con los requisitos del Reglamento sobre productos sanitarios (MDR) y el Reglamento sobre dispositivos médicos (IVDR), utilizando metodologías de búsqueda avanzadas. Las publicaciones de bases de datos de todo el mundo se examinan y analizan de forma sistemática para identificar la evidencia relevante que respalde la seguridad, el rendimiento y los beneficios clínicos de los dispositivos.
Protocolo de búsqueda Dispositivos Médicos y revisión sobre diagnósticos in vitro (IVD) y Dispositivos Médicos
- Identificación, recopilación y síntesis sistemáticas de la literatura científica
- Diseño y aplicación de protocolos de búsqueda bibliográfica conformes con el MDR y el IVDR
- Definición de las preguntas de investigación y de estrategias de búsqueda adaptadas al dispositivo.
- Identificación de palabras clave, creación de cadenas de búsqueda y selección de bases de datos (PubMed, Embase, Cochrane, etc.)
- Evaluación crítica de la evidencia clínica, de rendimiento y científica.
- Resumen de pruebas para la documentación reglamentaria.
- Elaboración de CER, CEP, PER y PEP
- Evaluación de las deficiencias en los datos de la documentación existente sobre CER, CEP, PEP y PER.
- Utilización de técnicas de búsqueda avanzadas para recopilar bibliografía relevante a nivel mundial

- Cumplimiento garantizado del MDR/IVDR
- Proceso de búsqueda bibliográfica estructurado, reproducible y fundamentado
- Estrategias de evidencia personalizadas y específicas para cada dispositivo
- Expertos clínicos y normativos altamente cualificados
- Capacidad del equipo ampliable; estrategias de recopilación de pruebas personalizadas y específicas para cada dispositivo
- Aportaciones interdisciplinarias en materia de regulación, medicina y clínica
- Asistencia End-to-end en la búsqueda, revisión y documentación End-to-end
- Mejora la credibilidad, la claridad y la preparación de las solicitudes reglamentarias

Preguntas Frecuentes (PF)
01. ¿Cuál es el objetivo de un protocolo de búsqueda Dispositivos Médicos en el marco del Reglamento sobre productos sanitarios (MDR) y el Reglamento sobre los productos sanitarios para diagnóstico in vitro ( EU MDR?
Un protocolo de búsqueda Dispositivos Médicos ofrece un enfoque estructurado, sistemático y transparente para identificar, evaluar y documentar la evidencia científica sobre un dispositivo o sus comparadores. Garantiza la reproducibilidad, minimiza el sesgo y permite a las autoridades reguladoras rastrear cómo se recopiló, evaluó y sintetizó la evidencia clínica o de rendimiento. En el marco EU MDR, dicho protocolo respalda las evaluaciones de seguridad, rendimiento y relación beneficio-riesgo, garantiza el cumplimiento normativo y constituye la base para una documentación de alta calidad y defendible a lo largo de todo el ciclo de vida del producto sanitario.
02. ¿Cómo influye el estado actual de la técnica en la evaluación clínica y del rendimiento?
El estado actual de la técnica (SOTA) representa el conocimiento científico y clínico reconocido en la actualidad para un tipo de dispositivo. Establece un punto de referencia en cuanto a la seguridad, el rendimiento y los resultados clínicos esperados. Determinar el SOTA mediante una revisión bibliográfica ayuda a contextualizar las afirmaciones sobre el dispositivo, facilita la selección de comparadores y orienta el análisis de la relación beneficio-riesgo, la planificación PMCF y las actualizaciones de la evidencia a lo largo del ciclo de vida del producto.
03. ¿En qué se diferencia una revisión bibliográfica MDR de una revisión sistemática tradicional?
Una revisión bibliográfica conforme al MDR/IVDR se diferencia de una revisión sistemática tradicional en que se centra en los aspectos normativos y está diseñada específicamente para respaldar el cumplimiento de los requisitos reglamentarios. Mientras que las revisiones sistemáticas tradicionales tienen como objetivo responder a preguntas de investigación científica y sirven a fines puramente académicos, una revisión bibliográfica del MDR evalúa la evidencia clínica y de rendimiento para demostrar la seguridad, el rendimiento y los perfiles de beneficio-riesgo de los productos sanitarios. Sigue una metodología estructurada y trazable con preguntas de investigación predefinidas, criterios de inclusión/exclusión y una evaluación crítica para producir documentación defendible y lista para auditorías con vistas a las presentaciones reglamentarias.
04. ¿Con qué frecuencia deben actualizarse las revisiones bibliográficas sobre productos sanitarios y productos para el diagnóstico in vitro?
La frecuencia de las actualizaciones depende del riesgo del dispositivo, la dinámica del mercado y la evolución de los datos disponibles. Los dispositivos de alto riesgo suelen requerir actualizaciones anuales, mientras que otros pueden seguir intervalos definidos. Las revisiones también deben actualizarse cuando surjan señales de seguridad significativas, nuevos datos clínicos, avances tecnológicos o cambios en las directrices, con el fin de mantener perfiles de beneficio-riesgo precisos.
05. ¿Qué papel desempeñan los criterios de inclusión y exclusión en las búsquedas bibliográficas sobre el Reglamento MDR y el Reglamento IVDR?
Los criterios de inclusión y exclusión garantizan que solo se seleccione evidencia relevante y de alta calidad. Mejoran la objetividad, reducen el sesgo de los revisores y garantizan la coherencia en la toma de decisiones. En el marco del MDR/IVDR, estos criterios deben estar predefinidos, justificados y alineados con las preguntas de investigación para mantener la trazabilidad y la defendibilidad normativa a lo largo de todo el proceso de evaluación.
06. ¿Por qué es fundamental la evaluación crítica en las revisiones bibliográficas relacionadas con el MDR y el IVDR?
La evaluación crítica analiza la calidad metodológica, la pertinencia y la fiabilidad de la evidencia incluida. Los marcos normativos del MDR y el IVDR hacen hincapié en la evaluación, ya que las autoridades reguladoras se basan en afirmaciones sobre la seguridad y el rendimiento que estén bien fundamentadas. Una evaluación rigurosa ayuda a distinguir los datos sólidos de los estudios menos sólidos y refuerza las conclusiones utilizadas en las evaluaciones comparativas de la evidencia (CER), las evaluaciones de la eficacia y la seguridad (PER), los informes de seguimiento poscomercialización (PMS) y los análisis de la relación beneficio-riesgo.
07. ¿En qué se diferencian los requisitos de búsqueda bibliográfica del MDR y del IVDR?
El Reglamento sobre productos sanitarios (MDR) se centra en la evaluación clínica, la justificación de la relación beneficio-riesgo y el rendimiento clínico, mientras que el Reglamento sobre productos sanitarios para diagnóstico in vitro (IVDR) hace hincapié en el rendimiento analítico, la validez científica y el rendimiento clínico en lo que respecta a la precisión diagnóstica. Las estrategias de revisión bibliográfica deben reflejar estas diferencias adaptando las preguntas de investigación, los conjuntos de datos y los marcos de evaluación a las distintas vías de evidencia que exige cada normativa.
08. ¿Qué bases de datos y fuentes de información se deben utilizar en las búsquedas bibliográficas que se ajusten a los requisitos del MDR y el IVDR?
Las autoridades reguladoras esperan que se utilicen múltiples bases de datos científicas, como PubMed, Embase y Cochrane, complementadas con bases de datos de vigilancia, registros de ensayos clínicos, guías y literatura gris pertinente. El uso de fuentes diversas garantiza una cobertura exhaustiva de la información clínica, de rendimiento y de seguridad necesaria para una evaluación sólida y una vigilancia continua.
09. ¿Por qué se considera a Freyr un socio de referencia en la búsqueda bibliográfica y la elaboración de protocolos?
Freyr es considerado un socio de referencia gracias a su profundo conocimiento de la normativa, su rigor científico y su cumplimiento sistemático de los requisitos de evidencia establecidos por el MDR y el IVDR. El equipo aplica principios de revisión sistemática, una metodología transparente y su experiencia en áreas terapéuticas para generar resultados sólidos y preparados para una auditoría. El enfoque de Freyr hace hincapié en la trazabilidad, la evaluación crítica y la comparación con los últimos avances, factores clave valorados por los organismos notificados y las autoridades reguladoras de todo el mundo.







