
Resumen del Registro de Dispositivos Médicos en Israel
La industria de dispositivos médicos de Israel está experimentando un crecimiento sostenido y una innovación, lo que la convierte en un centro para tecnologías sanitarias de vanguardia. El registro de dispositivos médicos es crucial para las empresas que ingresan a este mercado dinámico. Este resumen explora aspectos clave del proceso de registro en Israel, ofreciendo información sobre el marco reglamentario y los requisitos para llevar dispositivos médicos innovadores a la vanguardia del sector sanitario de Israel.
Autoridad Reglamentaria: La División de Dispositivos Médicos del Ministerio de Salud de Israel (AMAR).
Reglamento: Ley de Dispositivos Médicos/Equipos de 2012
Vía Reglamentaria: Registro de Productos
Representante autorizado local en Israel: Titular del registro en Israel (IRH)
Requisito de QMS: ISO 13485
Evaluación de datos técnicos: El Departamento de Dispositivos Médicos del Ministerio de Salud
Validez de la Licencia: Cinco (05) años
Formato de Presentación: En papel y electrónico
Traducción: Documentos Traducidos en Hebreo
Clasificación de Dispositivos
La Ley de Equipos Médicos y el Reglamento para el Registro de Equipos Médicos en Israel no especifican un sistema de clasificación de riesgos. En cambio, Israel alinea su clasificación de Dispositivos Médicos con los estándares internacionales, en particular los establecidos por los países del Grupo de Trabajo de Armonización Global (GHTF). Alternativamente, la clasificación de riesgo de un Dispositivo en un país reconocido se adopta para el registro en Israel. Este proceso de clasificación suele considerar el uso previsto, el nivel de riesgo y otros factores que pueden afectar la seguridad y eficacia de los Dispositivos Médicos.
Clases de Dispositivos Médicos
| Clase | Riesgo |
|---|---|
| Clase I | Baja |
| Clase II | Bajo-Medio |
| Clase III | Alta |
Cambios propuestos para las rutas de registro
Las modificaciones propuestas se aplican a los dispositivos de Clase I y Clase II, mientras que el sistema de registro para los dispositivos de Clase III permanece sin cambios.
- Los dispositivos de Clase I pueden registrarse inmediatamente mediante autodeclaración.
- Para los dispositivos de Clase II, si bien las declaraciones y los documentos técnicos son necesarios, AMAR puede agilizar el proceso a catorce (14) días para aquellos considerados de riesgo bajo-medio. Esto se aplica si el fabricante posee dos (02) autorizaciones de países reconocidos y proporciona seis (06) meses de datos de mercado. Alternativamente, para los dispositivos de Clase II que solo cuentan con la autorización 510(k) de la FDA de US y seis (06) meses de datos del mercado de US, el tiempo de procesamiento de AMAR se acelera a sesenta (60) días.
Representante Autorizado Local en Israel
Las empresas de dispositivos médicos con sede fuera de Israel deben designar un Titular de Registro Israelí (IRH) para facilitar el registro de sus productos para la venta dentro del país. El IRH actúa como representante local del fabricante y tiene la tarea de establecer contacto con el Ministerio de Salud para asegurar el cumplimiento de las regulaciones locales. Además, un IRH es responsable de establecer y mantener una presencia comercial en Israel, así como de obtener y conservar una licencia comercial válida.
Registro de Dispositivos Médicos
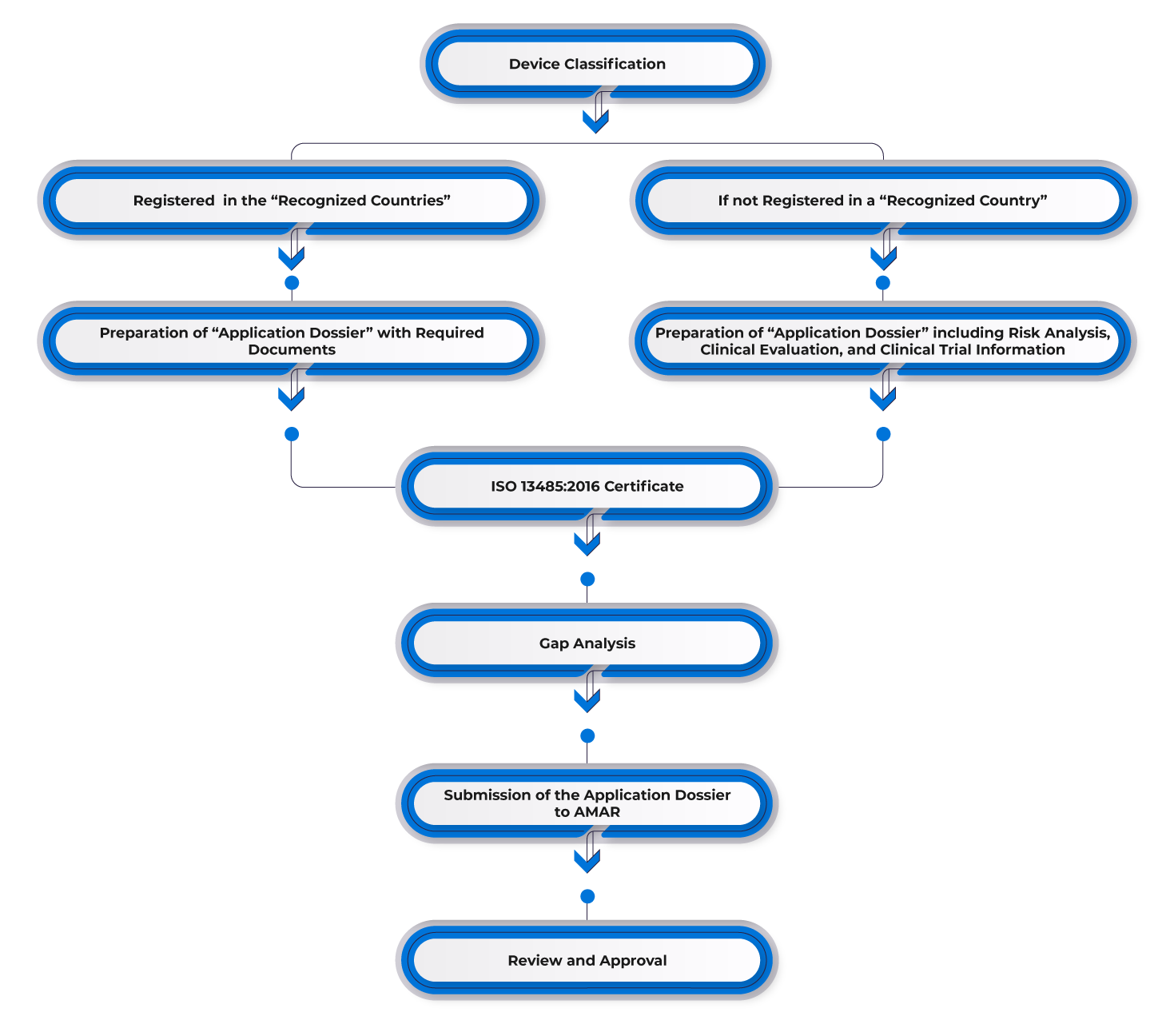
Para registrar un Dispositivo Médico en Israel, los fabricantes deben obtener una aprobación previa en uno de los mercados de referencia, como EE. UU., Europa, Australia, Canadá u otros mercados importantes. Los fabricantes con aprobaciones existentes en uno de los países de referencia pueden utilizar esta aprobación para el mercado israelí y nombrar un representante en el país. Posteriormente, deben presentar la documentación requerida, que incluye:
- FDA 510(k)/Carta de aprobación previa a la comercialización/CE.
- Certificado para Gobierno Extranjero (CFG)/Certificado de Libre Venta (CFS).
- Certificación ISO 13485 o cualquier otra certificación reconocida de Buenas Prácticas de Fabricación (BPF).
- Validación y Certificación del Instituto de Normas de Israel (Si es necesario).
Flujo del Proceso
Gestión del Ciclo de Vida del Dispositivo Post-Aprobación
Freyr ofrece soporte integral a fabricantes extranjeros en la gestión de todo el ciclo de vida de los Dispositivos Médicos en Israel, incluidas las actividades posteriores a la aprobación:
- Gestión de cambios post-aprobación, que aborda las modificaciones a las aprobaciones existentes de Dispositivos Médicos, como la adición de nuevas variantes, accesorios e indicaciones de uso.
- Mantenimiento de la certificación ISO 13485:2016 y CE.
- Renovación de licencias.
- Actuar como intermediario entre el Organismo Notificado (ON) y el fabricante.
Navegar por las complejidades de los organismos de autorización y cumplir con múltiples conjuntos de regulaciones para la aprobación de Dispositivos Médicos puede ser un desafío. Obtener aprobaciones de varios países del GHTF y adherirse a las regulaciones estatales requiere un conocimiento reglamentario profundo. Para los nuevos participantes en el mercado que se enfrentan a estas complejidades sin un socio reglamentario establecido, Freyr ofrece servicios reglamentarios End-to-End, simplificando el proceso de aprobación para Dispositivos Médicos en Israel.
Experiencia en el Registro de Dispositivos Médicos en Egipto
- Clasificación de Dispositivos Médicos en Israel.
- Titular de Registro en Israel (IRH).
- Registro de Dispositivos en Israel.
- Consultoría en Gestión de Riesgos ISO 14971:2019.
- Cumplimiento de la ISO 13485:2016.
- Revisión, compilación y presentación del expediente de diseño.
- Registro de Dispositivos Médicos a través del Sistema de Registro en Línea.
- Informe de Estrategia Reglamentaria de Dispositivos Médicos.
- Soporte de Pruebas - Biocompatibilidad, Seguridad Eléctrica, Mecánica y Rendimiento.
- Soporte de Cumplimiento de Etiquetado.
- Apoyo GMP.
- Soporte de Vigilancia Post-comercialización (PMS).
