

Resumen del Registro de Dispositivos Médicos en Nueva Zelanda
Los Dispositivos Médicos en Nueva Zelanda están regulados por la Autoridad de Seguridad de Medicamentos y Dispositivos Médicos de Nueva Zelanda (Medsafe) según el Reglamento de Medicamentos de 1984, la Ley de Medicamentos de 1981 y el Reglamento de Medicamentos (Base de Datos de Dispositivos Médicos) de 2003. Si bien la aprobación previa a la comercialización no es necesaria, es imprescindible registrar los productos en la base de datos del sistema de Notificación de Dispositivos Asistida por Web Electrónica (WAND) dentro de los 30 días posteriores al lanzamiento comercial. Medsafe puede solicitar documentación que demuestre la seguridad y eficacia, como la certificación de organismos reconocidos como un Organismo Notificado de la UE o Health Canada.
El equipo de expertos reglamentarios en Dispositivos Médicos de Freyr aporta una considerable experiencia en guiar a las empresas de Dispositivos Médicos a través del proceso de registro de Medsafe para Dispositivos Médicos en Nueva Zelanda.
![]()
Autoridad reglamentaria: Autoridad de Seguridad de Dispositivos Médicos (Medsafe)![]()
Reglamentación:El Reglamento de Medicamentos (Base de Datos de Dispositivos Médicos), 2003
Ley de Medicamentos de 1981
Reglamento de Medicamentos de 1984![]()
Ruta Reglamentaria: Sistema Electrónico de Notificación de Dispositivos Asistido por Web (WAND)![]()
Representante Autorizado: Patrocinador de Dispositivos Médicos![]()
Requisito de QMS: Certificación ISO 13485:2016![]()
Evaluación de Datos Técnicos: Autoridad de Seguridad de Dispositivos Médicos (Medsafe)![]()
Validez de la licencia: Los listados de dispositivos en Nueva Zelanda no caducan. Los dispositivos que se considere que representan una amenaza importante para el público pueden ser retirados del mercado.![]()
Requisitos de Etiquetado: Reglamento 12(4) del Reglamento de Medicamentos de 1984 y GHTF/SG1/N43:2005![]()
Formato de Presentación: Sistema Electrónico de Notificación de Dispositivos Asistido por Web (WAND)![]()
Idioma: Inglés
Clasificación de Dispositivos Médicos en Nueva Zelanda
Los Dispositivos Médicos en Nueva Zelanda se clasifican por riesgo en Clases I, IIa, IIb, III y AIMD de acuerdo con los criterios del Foro Internacional de Reguladores de Dispositivos Médicos (IMDRF). Esta clasificación afecta la cantidad de control reglamentario necesario. La clasificación se basa en características como el propósito previsto del Dispositivo Médico, la duración del contacto con el cuerpo, la invasividad y si es activo o inactivo. Los Dispositivos Médicos de clase superior están sujetos a una supervisión reglamentaria más estricta. Medsafe es la entidad reglamentaria en Nueva Zelanda que supervisa estas clasificaciones y regulaciones.
| Clasificación de Dispositivos Médicos de Medsafe, distinta de la clase de DIV | Riesgo |
|---|---|
| Clase I Básica | Riesgo bajo |
| Clase I de medición | Riesgo bajo |
| Clase I estéril | Riesgo bajo |
| Clase IIa | Riesgo bajo-medio |
| Clase IIb | Riesgo medio-alto |
| Clase III y Dispositivo Médico implantable activo (AIMD) | Riesgo elevado |
| Clasificación de IVD de Medsafe | Riesgo |
|---|---|
| A partir de julio de 2014, Medsafe no reconoce ningún sistema de clasificación de riesgos para los IVD. Todos los IVD notificados a WAND deben utilizar el código de clasificación de riesgos del IVD. El Director General de Salud autorizó la exención para los IVD de acuerdo con el Anexo 1, párrafo (i) del Reglamento de Medicamentos (Base de Datos de Dispositivos Médicos) de 2003. Pero los proveedores de IVD pueden notificar voluntariamente sus dispositivos a la base de datos. | |
Representante Autorizado/Patrocinador de Dispositivos Médicos
El Representante Autorizado es denominado Patrocinador y actúa como intermediario entre el fabricante y Medsafe. Los Patrocinadores actúan como representantes reglamentarios para los productos que se comercializan en Nueva Zelanda, presentando solicitudes WAND y actuando como el principal punto de contacto entre el fabricante y Medsafe para todos los asuntos relacionados con el producto. Además, Medsafe responsabiliza al Patrocinador de los esfuerzos de vigilancia.
Registro de Dispositivos Médicos en Nueva Zelanda
El registro de Dispositivos Médicos, el procedimiento de Nueva Zelanda y el procedimiento de listado WAND en Nueva Zelanda varían según la clase del dispositivo.
Dispositivos de Clase I- Se requiere una declaración de conformidad del fabricante para equipos de Clase I no estériles y no medidores; sin embargo, rara vez se presenta a un organismo reglamentario. En su lugar, el patrocinador (o proveedor) debe introducir los detalles del dispositivo en la base de datos Web Assisted Notification of Devices (WAND) como parte del proceso de notificación de Medsafe.
Dispositivos de otras clases
En Nueva Zelanda, los patrocinadores o proveedores son responsables de asegurar que los Dispositivos Médicos cumplan normas como la ISO 13485:2016. Normalmente no se requiere la presentación directa de una Declaración de Conformidad, una certificación de SGC o pruebas de fabricación a Medsafe. Sin embargo, conservar esta documentación es crucial para demostrar el cumplimiento si se solicita.
Medsafe prioriza la vigilancia post-comercialización sobre la aprobación detallada previa a la comercialización para los Dispositivos Médicos. Aunque las auditorías no se realizan de forma rutinaria durante la fase de notificación, Medsafe puede iniciarlas para dispositivos de mayor riesgo o tras actividades de vigilancia e informes de eventos adversos, garantizando la seguridad y el cumplimiento continuos.
Una vez que un Dispositivo es notificado a través de la base de datos WAND, puede comercializarse en Nueva Zelanda, siempre que el proveedor cumpla constantemente con las regulaciones de Medsafe. Esto exige un cumplimiento continuo, particularmente con los estándares de monitorización poscomercialización y notificación de incidentes. Los expertos en Dispositivos Médicos de Freyr apoyan los servicios relacionados con la navegación de estos requisitos reglamentarios, asegurando que las empresas mantengan el cumplimiento durante todo el ciclo de vida del producto.
Flujo del proceso
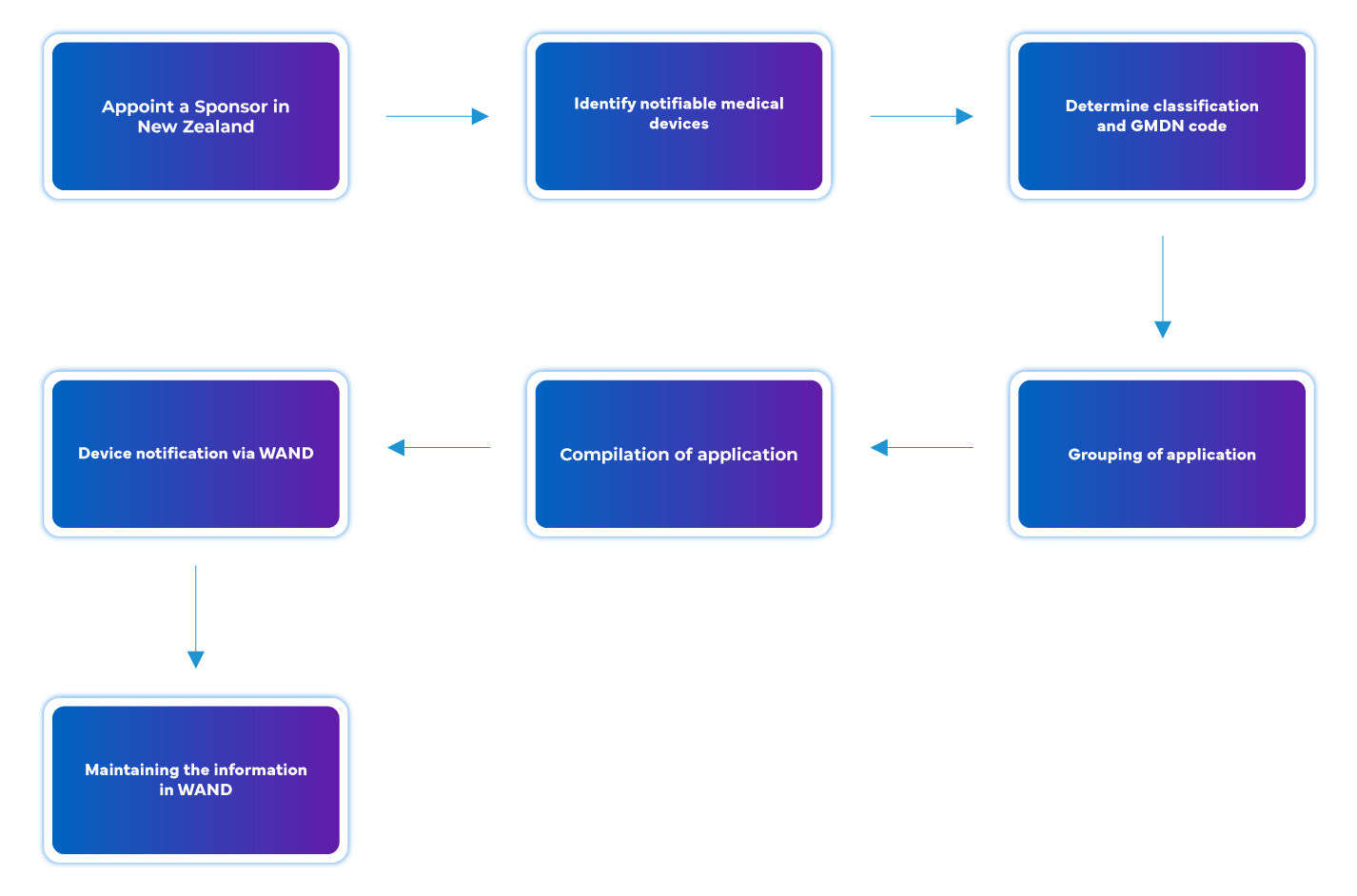
Gestión del ciclo de vida de dispositivos post-aprobación
Freyr apoya a fabricantes extranjeros en la gestión End-to-End del ciclo de vida de Dispositivos Médicos, incluyendo actividades posteriores a la aprobación mediante la notificación a las autoridades de Nueva Zelanda a través de WAND, tales como –
- Gestión de cambios post-aprobación: modificaciones a las aprobaciones existentes de Dispositivos Médicos, como la adición de nuevas variantes, accesorios; adición de nuevas indicaciones de uso, entre otros.
- Mantenimiento de las aprobaciones y el registro.
Equipado con un equipo de profesionales reglamentarios, Freyr ofrece un soporte integral a los fabricantes para mantener los estándares de calidad y seguridad requeridos para la aprobación en el mercado. Los especialistas en inteligencia reglamentaria de la firma supervisan meticulosamente las actualizaciones en las regulaciones, asegurando que los clientes estén bien informados sobre las acciones necesarias para mantener la conformidad de sus productos con los estándares actuales.
Resumen
| Riesgo | Clase de Dispositivo | Auditoría de SGC | Ruta reglamentaria | Plazos de Medsafe | Validez del Registro (años) |
|---|---|---|---|---|---|
| Riesgo bajo | Clase I Básica | Cumplimiento de la ISO 13485:2016 Nota: Medsafe no exige auditorías de SGC, pero recomienda encarecidamente seguir la ISO 13485:2016 para la calidad y la seguridad. Medsafe tiene autoridad para realizar auditorías de SGC para cualquier clase de dispositivo si surgen problemas de seguridad o calidad. | Registro WAND (Notificación) | 1 semana |
Sin fechas de caducidad |
| Riesgo bajo | Clase I de medición | Registro WAND (Notificación) | |||
| Riesgo bajo | Clase I estéril | Registro WAND (Notificación) | |||
| Riesgo bajo-medio | Clase IIa | Registro WAND (Notificación) | |||
| Riesgo medio-alto | Clase IIb | Registro WAND (Notificación) | |||
| Riesgo elevado | Clase III | Registro WAND (Notificación) |
Nota: Según la legislación actual, los registros de dispositivos en Nueva Zelanda no caducan, pero los dispositivos que se consideren que representan un riesgo inaceptable para el público pueden ser retirados del mercado. Sin embargo, la legislación actual podría ser revisada para 2026/2027.
Experiencia de Freyr
- Soporte End-to-End para el registro de Dispositivos Médicos.
- Soporte de LR
- Registro WAND
- Apoyo en etiquetado
- Gestión de Cambios Post-aprobación
- Transferencia de licencia
- Servicios de presentación y enlace con WAND