


Descripción general del registro de Dispositivos Médicos en Turquía
El mercado turco de Dispositivos Médicos ha experimentado un crecimiento significativo y constante durante la última década. A partir de 2021, el Registro de Dispositivos Médicos de Turquía requiere cumplir con el Reglamento de Dispositivos Médicos de la UE (EU MDR) 2017/745 y el Reglamento de Dispositivos Médicos de Diagnóstico In Vitro (IVDR) 2017/746. Esto ha impulsado el comercio internacional, llevando a varias empresas globales a lanzar sus Dispositivos Médicos en el país.
![]()
Autoridad reglamentaria: Agencia Turca de Medicamentos y Dispositivos Médicos (TITCK)![]()
Reglamentación: Reglamentaciones de Dispositivos Médicos (MDR) 2017/745, Reglamentaciones de Dispositivos Médicos para Diagnóstico In Vitro 2017/746![]()
Ruta Reglamentaria: El marcado CE es obligatorio, seguido de Registro/Notificación en el Sistema de Seguimiento de Productos (UTS)![]()
Representante local en Turquía![]()
Requisito de QMS: ISO 13485:2016![]()
Evaluación de Datos Técnicos: Organismo Notificado para el Marcado CE![]()
Validez de la licencia: Ilimitada![]()
Formato de Presentación: En papel![]()
Traducción: Documentos Traducidos al Turco
Clasificación de Dispositivos
Turquía sigue la misma clasificación de dispositivos médicos que la establecida en el EU MDR y el IVDR. Determinar la clasificación del dispositivo puede ser un desafío, por lo que contar con el respaldo de un consultor reglamentario experimentado es crucial.
Clases de Dispositivos Médicos
| Clase | Riesgo |
|---|---|
| Clase I | Baja |
| Clase IIa | Moderado |
| Clase IIb | Moderado a Alto |
| Clase III | Alta |
Clases de dispositivos de diagnóstico in vitro
| Clase | Riesgo |
|---|---|
| Clase A | Baja |
| Clase B | Moderado |
| Clase C | Moderado a Alto |
| Clase D | Alta |
Registro de Dispositivos Médicos
El marcado CE es una conformidad que los fabricantes deben cumplir para comercializar su dispositivo en el mercado de Turquía. El marcado CE se emite a través de una evaluación de conformidad realizada por el organismo notificado. Ahora, Turquía está autorizada para designar organismos notificados de acuerdo con la EU MDR y la IVDR.
Las empresas deben registrarse en el Sistema Central de Registro (MERSIS) y registrar el dispositivo en el Sistema de Seguimiento de Productos (UTS).
Flujo del proceso
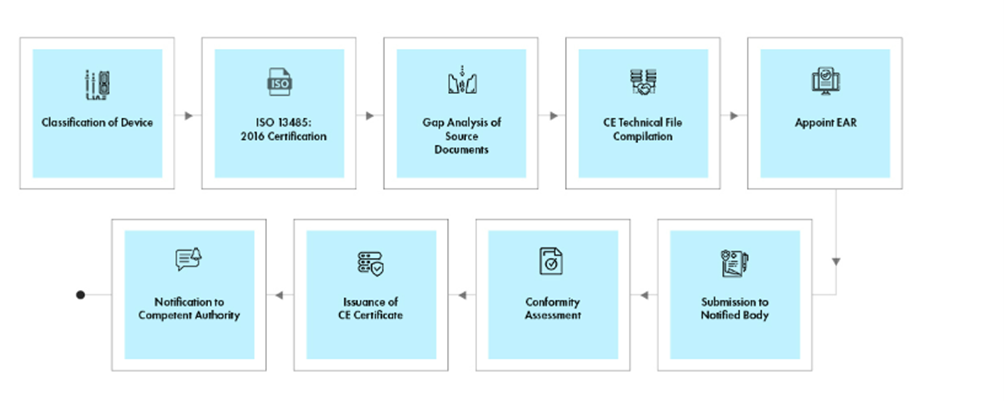
Gestión del Ciclo de Vida del Dispositivo Post-Aprobación
Freyr apoya a fabricantes extranjeros en la gestión End-to-End del ciclo de vida de Dispositivos Médicos, incluyendo actividades posteriores a la aprobación, tales como:
- Gestión de cambios post-aprobación: modificaciones a las aprobaciones existentes de Dispositivos Médicos, como la adición de nuevas variantes, accesorios; adición de nuevas indicaciones de uso, entre otros.
- Mantenimiento de la certificación ISO 13485:2016 y CE
- Renovación de licencias
- Coordinación entre el organismo notificado y el fabricante
Con la participación de varios organismos de autorización, los fabricantes extranjeros deben cumplir con múltiples conjuntos de regulaciones en cada proceso individual para la aprobación de dispositivos. La obtención del marcado CE y la posterior adhesión a las regulaciones estatales requieren un amplio conocimiento reglamentario. A veces, sin un socio reglamentario probado, navegar por todos los requisitos de los dispositivos puede ser un desafío para los nuevos participantes en el mercado. Para ayudar a los fabricantes, Freyr ofrece servicios reglamentarios End-to-End para agilizar las aprobaciones de Dispositivos Médicos.
Experiencia de Freyr
- Clasificación Europea de Dispositivos Médicos
- Apoyo al Representante Autorizado Europeo (EAR)
- Registro de Dispositivos y Notificación de Productos en Turquía
- Consulta sobre gestión de riesgos ISO 14971:2019
- Cumplimiento de ISO 13485:2016
- Revisión, compilación y presentación del expediente técnico CE/dossier de diseño
- Soporte para la transición de EU MDR
- Apoyo para la Transición al IVDR de la UE
- Clinical Evaluation Reports (CER) para Dispositivos Médicos.
- Informes de Evaluación de Desempeño (PER) para Dispositivos de Diagnóstico In Vitro
- Notificación/Registro de Dispositivos Médicos a través del Sistema de Registro en Línea
- Informe de estrategia reglamentaria de Dispositivos Médicos
- Soporte de pruebas: biocompatibilidad, seguridad eléctrica, mecánica y rendimiento
- Soporte para el cumplimiento del etiquetado
- Soporte GMP
- Apoyo en la Vigilancia Post-Comercialización
