3 min de lectura
BIMO significa Bioresearch Monitoring (Monitorización de Biorinvestigación), un programa de inspección in situ y auditorías de datos para monitorizar todos los aspectos de la realización y la notificación de la investigación regulada por la Administración de Alimentos y Medicamentos de US (FDA). El programa se estableció en 1977 después de que se identificara la necesidad de auditar los sitios de investigación clínica. El objetivo principal de este programa es garantizar la calidad y la integridad de los datos presentados para las aprobaciones de nuevos productos y las solicitudes de comercialización. Además, este programa también protege los derechos y el bienestar de los sujetos humanos y animales involucrados en la investigación regulada por la FDA.
Los objetivos clave del Programa BIMO
Anualmente, se realizan más de 1000 inspecciones. Los objetivos principales cubiertos por el programa BIMO son:
- Auditar datos clínicos
- Inspección de investigación clínica en curso
- Inspección de laboratorios no clínicos.
- Inspección de Juntas de Revisión Institucional (IRB)
¿Qué productos están sujetos a la Auditoría BIMO?
El BIMO es aplicable a medicamentos, productos biológicos, Dispositivos Médicos, productos alimenticios, productos de tabaco y productos veterinarios. El programa de cumplimiento es supervisado por los seis (06) centros de productos de la FDA: Center for Biologics Evaluation and Research (CBER), Center for Devices and Radiological Health (CDRH), Center for Drug Evaluation and Research (CDER), Center for Food Safety and Applied Nutrition (CFSAN), Center for Tobacco Products (CTP) y Center for Veterinary Medicine (CVM).
¿Qué empresas están sujetas a la Auditoría BIMO?
Tanto las empresas nacionales como las internacionales que realizan o se encuentran dentro de alguna de las siguientes actividades están sujetas a los requisitos de la Monitorización de Bionvestigación -
- Laboratorios de pruebas no clínicas para el cumplimiento de las Buenas Prácticas de Laboratorio (GLP).
- Investigadores clínicos para el cumplimiento de las Buenas Prácticas Clínicas (GCP)
- Patrocinadores
- Organizaciones de Investigación por Contrato (CROs)
- Monitores de ensayos clínicos.
- Instalaciones de bioequivalencia in vivo
- Comités de Revisión Institucional (IRBs)
¿Qué programas de cumplimiento están incluidos en el Programa BIMO?
La US FDA puede realizar una auditoría BIMO en cualquier momento a través de los siete (07) programas de cumplimiento multicéntricos. Estos siete programas de cumplimiento multicéntricos se implementan a través de –
- Inspección de Investigador Clínico (CI) e Investigador Patrocinador (SI).
- Inspección del Comité de Ética en Investigación (IRB)
- Inspección de la Organización de Investigación por Contrato/Patrocinador/Monitor (CRO/S/M)
- Inspección de Buenas Prácticas de Laboratorio (BPL)
- Inspección de Bioequivalencia-Biodisponibilidad (BEQ)
- Inspección de informes de experiencias adversas a medicamentos (PADE) de postcomercialización
- Inspección de informes de la Estrategia de Evaluación y Mitigación de Riesgos (REMS)
Cada uno de estos programas describe un alcance detallado de revisión o inspección que se realizará para garantizar el cumplimiento con la FDA.
¿Qué reglamentos son aplicables a la Auditoría BIMO?
Los reglamentos - 21 CFR 50 - Protección de Sujetos Humanos, 21 CFR 54 - Divulgación Financiera, 21 CFR 56 - IRBs, 21 CFR 58 - Buenas Prácticas de Laboratorio para laboratorios no clínicos, 21 CFR 809 - Productos de Diagnóstico In Vitro, y 21 CFR 812 - Exención para Dispositivos en Investigación son aplicables a la Auditoría BIMO.
¿Cuántas auditorías se realizan anualmente bajo el Programa BIMO?
El número de auditorías BIMO realizadas por la FDA de US varía cada año. En los últimos años, el número de inspecciones in situ ha disminuido debido al inicio de la pandemia de COVID-19, y la FDA tuvo que suspender toda la vigilancia in situ de los estudios clínicos. Solo se estaban monitoreando estudios clínicos críticos y cruciales específicos.
Las “Evaluaciones Reglamentarias Remotas” (RRAs) se introdujeron durante la pandemia de COVID-19 para monitorear de forma remota la investigación reglamentada. Las RRAs se realizan mediante videoconferencias y son una iniciativa voluntaria para evaluar de forma remota los datos y procesos. Sin embargo, cabe señalar que las RRAs no son equivalentes ni una alternativa a la inspección in situ, sino que son simplemente un procedimiento que evolucionó debido a la pandemia de COVID-19.
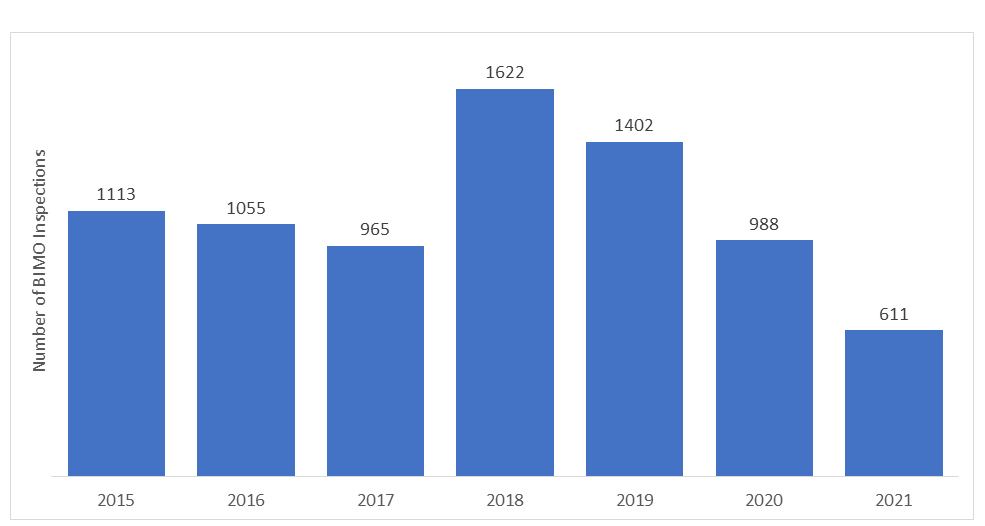
*Los datos representados para los años 2020 y 2021 no incluyen inspecciones de RRA
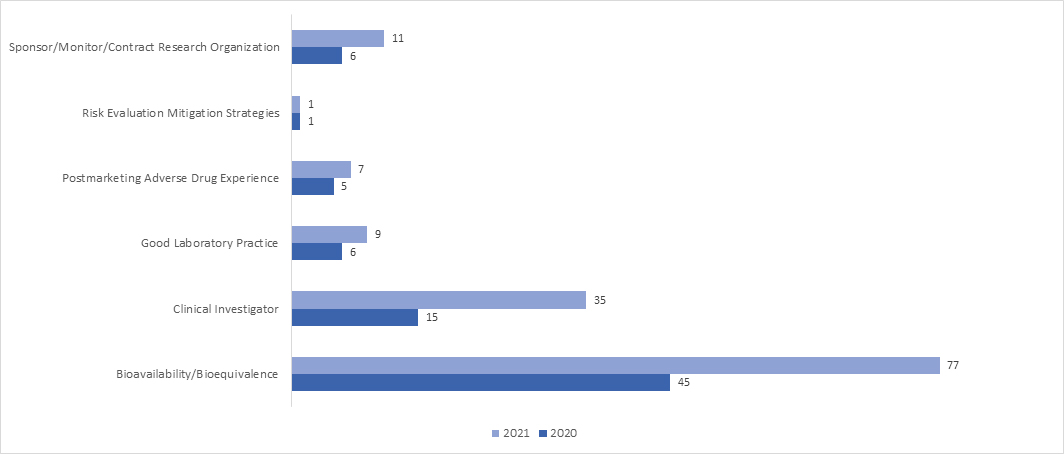
¿Cuántas Evaluaciones Reglamentarias Remotas (RRAs) se realizaron durante la pandemia de COVID-19 bajo el Programa BIMO?
En 2021, la adopción de las inspecciones RRA aumentó significativamente en todos los programas. En abril de 2021, la FDA publicó un documento de orientación sobre «Evaluaciones Interactivas Remotas de Instalaciones de Fabricación de Medicamentos y Monitoreo de Bionvestigación Durante la Emergencia de Salud Pública de COVID-19 - Guía para la Industria», que proporciona información exhaustiva sobre el proceso de la FDA para llevar a cabo las RRA.
¿Cuáles son los posibles resultados de una Auditoría BIMO?
Durante la auditoría BIMO, la US FDA puede decidir tomar cualquiera de las acciones que se enumeran a continuación, basándose en el cumplimiento –
1. No se requiere ninguna acción (NAI)
NAI es aplicable cuando el inspector de campo de la FDA no ha identificado ninguna práctica objetable o solo problemas menores para los cuales no se justifica una acción adicional.
2. Acción Voluntaria Indicada (VAI)
VAI es aplicable cuando se han identificado prácticas objetables, pero estas no son significativas.
3. Acción oficial indicada (OAI).
OAI es aplicable cuando se identifican prácticas objetables que comprometen la integridad de los datos y/o los derechos de los sujetos humanos.
¿Cuáles son las no conformidades más comunes detectadas en una Auditoría BIMO?
Algunas de las no conformidades más comunes observadas durante la Auditoría BIMO son –
- No llevar un seguimiento adecuado de los registros
- Incumplimiento en relación con el plan de investigación.
- Incumplimiento de las regulaciones
- Fallo en la supervisión de los protocolos
- Protección inadecuada del sujeto
- Responsabilidad inadecuada del producto que se está investigando
La auditoría BIMO es crucial para cualquier desarrollador o fabricante de nuevos Dispositivos Médicos y tecnologías que planee lanzar su dispositivo en el mercado de US. Cumplir con las regulaciones y directrices para evitar cualquiera de los inconvenientes descritos es muy importante.
¿Necesita ayuda con las inspecciones de auditoría BIMO? Contacte con Freyr. Manténgase informado. Manténgase conforme.