

3 min de lectura
Realizar un ensayo clínico es un paso importante en el desarrollo de un producto farmacéutico o un Dispositivo Médico. Se requiere una Solicitud de Ensayo Clínico (CTA) para que las empresas farmacéuticas o los patrocinadores obtengan la aprobación de las autoridades reglamentarias para iniciar estudios en humanos. A medida que la investigación clínica se vuelve más global y compleja, comprender las prácticas internacionales en evolución, los pasos críticos para la presentación y las ventajas que ofrece un socio o socios de Asuntos Regulatorios es crucial tanto para los patrocinadores como para las organizaciones de investigación.
Solicitud de Ensayo Clínico (CTA) y sus Secciones:
Una Solicitud de Ensayo Clínico (CTA) es el expediente reglamentario que los patrocinadores deben presentar a las autoridades reglamentarias nacionales (NRAs) antes de iniciar un estudio clínico con participantes humanos. Las CTA son obligatorias para la mayoría de los ensayos intervencionales de medicamentos, dispositivos o productos biológicos. Aseguran que el ensayo sea científicamente sólido, éticamente apropiado y esté alineado con las directrices locales e internacionales, como los estándares de Buenas Prácticas Clínicas (GCP) del Consejo Internacional de Armonización (ICH) y los requisitos nacionales.
Las secciones clave en una CTA típica incluyen (entre otras):
- Carta de presentación y formularios de solicitud
- Protocolo de estudio clínico y manual del investigador
- Expediente del producto y evidencia de Buenas Prácticas de Fabricación (GMP)
- Aprobaciones de ética y del Comité de Revisión Institucional (IRB)
- Documentación de seguro y protección del sujeto
- Plan de monitoreo de seguridad de datos y declaraciones financieras
Prácticas globales en las presentaciones de CTA
Los procesos y requisitos de CTA varían entre regiones, pero están surgiendo varios principios comunes:
1. Armonización de formatos y estándares
- Muchos países utilizan el Documento Técnico Común (CTD) del ICH, lo que agiliza las presentaciones de estudios multinacionales. Su estructura modular permite la revisión paralela y simplifica las actualizaciones.
- Las adaptaciones locales son comunes, como en las regiones de Asia-Pacífico y el Pacífico Occidental, donde los países suelen combinar el CTD con formularios nacionales, comentarios de revisión, necesidades lingüísticas o estudios puente.
2. Consulta reglamentaria
- Se fomenta la colaboración temprana con las agencias —como la FDA de US, las autoridades nacionales de la UE y las agencias de Japón, China y los principales mercados emergentes—, especialmente para los ensayos clínicos globales y multirregionales (MRCT). Las reuniones de asesoramiento científico reducen los rechazos futuros y orientan la optimización del protocolo.
3. Vías colaborativas y aceleradas
- Las agencias aceptan cada vez más la «confianza» o las evaluaciones colaborativas, donde aprovechan las revisiones o aprobaciones de autoridades reglamentarias estrictas, lo que aumenta la velocidad y la coherencia.
- Puede haber opciones de revisión acelerada o prioritaria, especialmente durante emergencias sanitarias o para terapias innovadoras.
4. Integración de la revisión ética
- En la UE y en muchas otras regiones, las revisiones éticas y reglamentarias pueden realizarse en paralelo o a través de plataformas coordinadas para evitar presentaciones separadas y acortar los tiempos de inicio.
5. Transparencia y publicación
- Ahora es estándar registrar los ensayos en bases de datos reconocidas antes de la aprobación, y muchos países informan públicamente sobre el estado de aprobación de las CTA, lo que contribuye a la transparencia global y a las mejores prácticas.
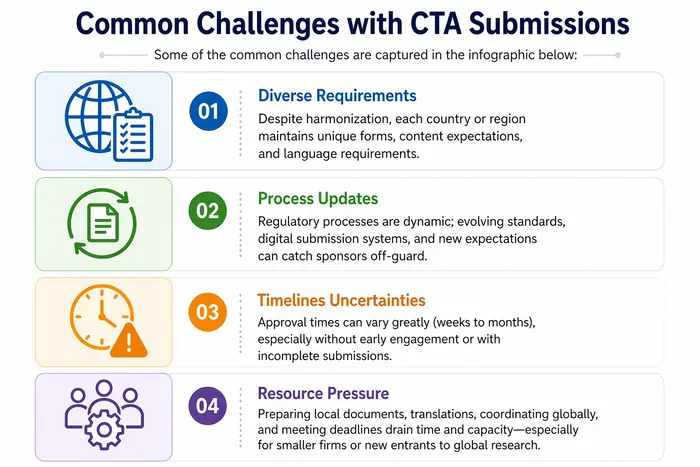
¿Cómo puede ayudar un socio como Freyr?
Un socio de confianza en Asuntos Regulatorios (RA) como Freyr proporciona un apoyo integral y mitigación de riesgos durante todo el proceso de CTA. He aquí por qué contar con un experto así prepara a los patrocinadores para el éxito:
1. Global Regulatory intelligence actualizada
- Monitoreamos continuamente los cambios reglamentarios, las interpretaciones regionales y las prácticas de presentación en todo el mundo. Esto asegura que cada CTA se elabore según los requisitos más actuales, minimizando consultas, rechazos y costosos retrasos en los ensayos.
2. Preparación y revisión de expedientes
- Los especialistas de Freyr desarrollan y compilan las CTA utilizando los últimos formatos ICH CTD y plantillas nacionales personalizadas. Coordinan las traducciones de documentos, organizan las respuestas a los expedientes y garantizan una calidad uniforme y el cumplimiento de las GCP para cada presentación.
3. Consultoría Reglamentaria y Enlace con Agencias
- Freyr puede representar a los patrocinadores en reuniones de asesoramiento científico previas a la CTA, gestionar las preguntas de las agencias después de la presentación y apoyar las comunicaciones tanto con las autoridades como con los Comités de Ética.
4. Gestión Global de Proyectos
- Con equipos multiculturales experimentados y herramientas sólidas de gestión de proyectos, Freyr coordina presentaciones simultáneas, realiza un seguimiento del progreso país por país e impulsa la alineación para los MRCT (Ensayos Clínicos Multirregionales).
5. Estrategia de Confianza y Revisión Acelerada
- Freyr ayuda a los patrocinadores a aprovechar los marcos de confianza, preparar expedientes abreviados para revisiones exentas o aceleradas, y alinear los datos de presentación para una máxima aceptación reglamentaria.
6. Preparación para Auditorías y Control de Cambios
- El socio garantiza que la documentación y los procedimientos de los patrocinadores cumplan con los estándares de auditoría, facilitando una rápida aprobación reglamentaria e inspecciones sin problemas.
Conclusión
La presentación de la CTA es una puerta de entrada compleja pero esencial para la investigación clínica, con requisitos globales y específicos de cada región. Las tendencias globales incluyen la armonización del CTD, la revisión ética y reglamentaria paralela, los mecanismos de confianza y las nuevas normas de transparencia. Los patrocinadores se enfrentan a desafíos importantes para cumplir con requisitos diversos, mantenerse al día con los cambios reglamentarios y gestionar los plazos globales. En tal escenario, un socio de Asuntos Regulatorios como Freyr proporciona experiencia End-to-End en inteligencia reglamentaria, preparación de expedientes, interacciones con agencias, vías aceleradas y preparación para auditorías, aumentando sustancialmente las probabilidades de iniciar ensayos clínicos de manera oportuna y exitosa.









