Conformità alle norme in materia di registrazione e regolamentazione dei dispositivi medici nell'Unione Europea
L'Unione Europea rappresenta uno dei mercati dei dispositivi medici più regolamentati e commercialmente attraenti al mondo. Con member states 27 member states, EEA 3 EEA e la Turchia, il mercato dell'Unione opera nell'ambito di un ecosistema normativo armonizzato definito dal EU MDR 2017/745) per i dispositivi medici e dal regolamento UE IVDR (2017/746) per i dispositivi diagnostici in vitro; la regione garantisce standard uniformi di sicurezza e prestazioni in tutte le categorie di prodotti. Per immettere legalmente un dispositivo sul mercato dell'Unione Europea, produttori ottenere il marchio CE, che conferma la conformità alla normativa applicabile in base alla classificazione di rischio del dispositivo e al percorso di valutazione della conformità. Il mancato rispetto di questi requisiti può comportare gravi conseguenze, tra cui il richiamo del prodotto, il sequestro in dogana, la sospensione della certificazione o la completa perdita dell'accesso al mercato.
Ai sensi dell'attuale regime normativo:
- La marcatura CE è obbligatoria per tutti i dispositivi medici e i dispositivi diagnostici in vitro prima che possano essere immessi sul mercato.
- La registrazione dei dispositivi su EUDAMED viene introdotta in più fasi e diversi moduli sono già attivi e di uso obbligatorio.
- La nomina di un rappresentante autorizzato europeo (EAR) è obbligatoria per tutti i produttori con sede al di fuori dell'UE, delEEA.
- Le norme di classificazione più rigorose previste dal regolamento MDR/IVDR richiedono una documentazione tecnica solida, prove cliniche o di prestazione e un monitoraggio continuo durante l'intero ciclo di vita.
Freyr affianca produttori questo percorso, garantendo loro un ingresso agevole e conforme al mercato dell'Unione. Dall'elaborazione di una strategia per la marcatura CE e la redazione della documentazione tecnica alla gestione delle richieste presso gli organismi notificati e al ruolo di rappresentante autorizzato nell'UE, Freyr assicura la piena conformità alle normative MDR/IVDR e un accesso senza ostacoli a tutti member states dell'UE,EEA e alla Turchia.
Procedura dettagliata per la conformità alle normative UE
L'immissione sul mercato dell'UE di un dispositivo medico o di un dispositivo diagnostico in vitro (IVD) prevede diverse fasi ben definite. Ecco come Freyr gestisce l'intero processo per voi:
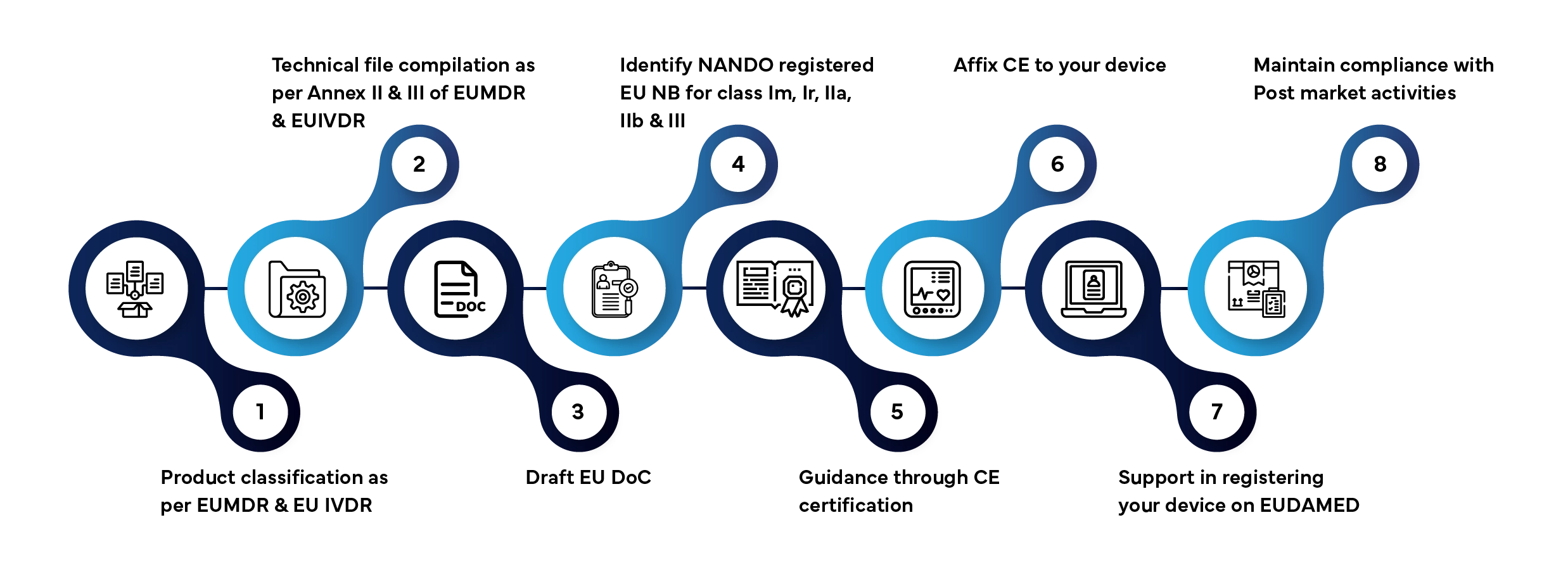
Tempi di elaborazione standard: da 3 a 12 mesi, a seconda della classe del dispositivo e della completezza della documentazione.
Principali offerte di Freyr Medical Device nell'Unione Europea
- Strategia normativa e transizione verso l'MDR e l'IVDR – Elaboriamo piani d'azione end-to-end e su misura per EU MDR l'IVDR EU MDR , garantendo una transizione agevole dai regimi previsti dalle direttive e l'allineamento con gli attuali requisiti dell'UE.
- Documentazione tecnica e valutazione della conformità – Il nostro team fornisce assistenza nello sviluppo, nella revisione e nella presentazione dei vostri fascicoli tecnici e dei dossier di progettazione, vi affianca nei rapporti con gli organismi notificati e gestisce il marchio CE, fornendo supporto per i test di sicurezza e di prestazione dei dispositivi e per i percorsi di conformità.
- Valutazione clinica e delle prestazioni: Freyr offre un servizio di redazione specialistica di CER, PER, piani PMCF, PSUR, SVR, CPR per dispositivi diagnostici in vitro, documentazione sui rischi e contenuti relativi alla valutazione biologica, garantendo chiarezza tecnica e accuratezza normativa per tutte le classi di dispositivi.
- Assistenza per la registrazione UDI ed EUDAMED – Garantiamo la conformità del vostro sistema di identificazione univoca dei dispositivi (UDI) e vi assistiamo nella registrazione nella banca dati europea dei dispositivi medici EUDAMED e nella gestione del ciclo di vita associata.
- Rappresentante autorizzato europeo (EAR) e rappresentanza locale – Per produttori EEA, operiamo in qualità di vostro rappresentante autorizzato europeo (EAR) e forniamo assistenza in materia di conformità normativa a livello locale in tutti member states dell'UE.
- Sorveglianza post-commercializzazione (PMS) – Freyr vi assiste nella definizione, nell'implementazione e nella gestione dei sistemi di sorveglianza post-commercializzazione, tra cui il Piano di sorveglianza post-commercializzazione (PMSP), il rapporto di sorveglianza post-commercializzazione (PMSR), il rapporto periodico di aggiornamento sulla sicurezza (PSUR), i piani di follow-up clinico/prestazionale post-commercializzazione (PMCF), la segnalazione di vigilanza, le FSCAs, le FSNs e le CAPAs, garantendo il mantenimento continuo del marchio CE e un accesso sostenibile al mercato con la conformità del ciclo di vita del prodotto.
- Registrazione dei dispositivi CE: Freyr gestisce l'intero iter di registrazione per il marchio CE nell'UE, fornendo assistenza nella valutazione della conformità, preparando le documentazioni conformi, interagendo con gli organismi notificati e garantendo approvazioni tempestive per tutte le categorie di dispositivi medici e di diagnostica in vitro.
- Supporto per i sistemi di gestione della qualità: forniamo assistenza nell'implementazione e nella gestione di sistemi di gestione della qualità ISO 13485, in linea con i requisiti di qualità e sicurezza previsti dal EU MDR e con le aspettative degli organismi notificati.
- Conformità dell'etichettatura: il nostro team garantisce che l'etichettatura, le istruzioni per l'uso, il packaging e i simboli siano conformi ai requisiti del MDR/IVDR e alle norme multilingue dell'UE, assicurando coerenza e conformità in tutti member states 27 member states dell'Unione Europea.
Servizi offerti dal Rappresentante autorizzato nell'UE (EAR)

Registrazione dei dispositivi presso le autorità dell'Unione Europea
Per produttori extra-UE, Freyr funge da rappresentante autorizzato nell'Unione europea (EAR) in conformità con i requisiti del MDR/IVDR. In qualità di entità con sede in Germania, Freyr fornisce assistenza nelle attività di registrazione dei dispositivi presso l'autorità competente tedesca di riferimento e conserva la documentazione normativa richiesta. Per Member States altri Member States dell'UE, Freyr fornisce assistenza normativa, ove applicabile, per garantire la conformità dei dispositivi e agevolarne l'immissione sul mercato dell'Unione.

Documentazione e garanzia di conformità
I nostri esperti in materia normativa verificano che la vostra Dichiarazione di conformità (DoC), i certificati CE e i fascicoli tecnici siano completi, aggiornati e conformi al MDR/IVDR. Freyr garantisce la piena preparazione per la valutazione di conformità e la presentazione delle richieste di marcatura CE.

Rispondere alle richieste delle autorità competenti
Se necessario, Freyr gestisce per vostro conto tutte le comunicazioni dirette e le richieste di chiarimenti da parte delle autorità competenti dell'UE o degli organismi notificati, garantendo risposte tempestive e accurate e contribuendo a evitare ritardi nelle approvazioni o nelle verifiche normative successive all'immissione sul mercato.

Vigilanza e comunicazione degli incidenti
In qualità di vostro EAR, Freyr funge da referente principale per le comunicazioni relative alla sicurezza. Se e quando necessario, coordiniamo le notifiche di incidenti, le azioni correttive di sicurezza sul campo (FSCA) e le segnalazioni di vigilanza tra il produttore, gli operatori sanitari e le autorità, al fine di garantire una risposta adeguata e la conformità alle normative.

Preparazione alle ispezioni e agli audit
Freyr conserva tutta la documentazione, la corrispondenza e i registri obbligatori ai fini delle verifiche e delle ispezioni da parte delle autorità. Il nostro team garantisce che la documentazione tecnica, l'etichettatura e i registri post-commercializzazione siano prontamente disponibili e pienamente conformi ai requisiti previsti dal MDR/IVDR.
Perché collaborare con Freyr?
- Competenza End-to-end , che spazia dalla registrazione pre-commercializzazione alla sorveglianza post-commercializzazione, gestendo ogni fase del processo di conformità.
- Una comprovata esperienza con oltre 2000 registrazioni di dispositivi completate con successo in diverse categorie.
- Una forte presenza nell'Unione Europea con sede in Germania (EAR), affiancata da esperti in materia normativa con sede a Reading e supportata dai team operativi globali di Freyr.
- Una pianificazione personalizzata della transizione che offre un supporto strategico per ottenere la certificazione CE in modo agevole ed economico.
- Team di consegna multiregionali con supporto sul posto grazie alle attività di Freyr nell'Unione Europea con sede in Germania.
- Comprovata esperienza nella messa a norma MDR/IVDR e nella transizione dei dispositivi preesistenti
- Gestione trasparente del progetto con comunicazione diretta con l'organismo notificato

Celebrazione del successo dei clienti
Dispositivi Medici
Supporto per la registrazione e LR
Globale
Freyr è stato un partner indispensabile per raggiungere una rapida scalabilità globale per la nostra attività di Software as a Medical Device (SaMD). Per una startup, acquisire competenze nelle normative mondiali è proibitivo in termini di costi. I prezzi competitivi e i servizi su misura di Freyr ci hanno permesso di ottenere tale competenza a una frazione del costo delle risorse a tempo pieno. La reattività e l'adattabilità del loro team alle priorità del progetto hanno notevolmente facilitato i nostri progressi. Raccomandiamo Freyr a qualsiasi azienda che cerchi una guida e un supporto esperti nel settore normativo dei dispositivi medici.
Arie Henkin
VP - Qualità e Affari Regolatori, con sede in Australia, Azienda leader nel settore SaMD
Dispositivi Medici
Servizi di Rappresentanza Svizzera
Giappone e Svizzera
Apprezzo sinceramente il tempo trascorso a lavorare con Freyr e li considero una risorsa davvero preziosa e un'estensione del mio team. Sono affidabili e precisi, e i loro prezzi sono competitivi. Inoltre, non esiterò a collaborare nuovamente con Freyr.
Darren Mansell
Responsabile Affari Regolatori, con sede nel Regno Unito, Azienda globale di progettazione e produzione di dispositivi medici
Dispositivi Medici
Registrazione e Servizi AR
Malesia e Indonesia
Freyr fornisce un servizio affidabile con esperienza in molti paesi. Posso fare affidamento su Freyr per fornire le informazioni necessarie a prendere una decisione informata prima di stipulare un accordo formale sull'ambito di lavoro. Una volta avviato un progetto, il team Freyr agisce professionalmente per eseguire il lavoro con un'ottima comunicazione dei progressi.