
Rapporto di valutazione clinica (CER) per dispositivi medici Panoramica
Qualsiasi dispositivo destinato alla commercializzazione nell'Unione Europea (UE) deve recare il marchio CE. In conformità con il regolamento EU MDR , i requisiti relativi alla relazione di valutazione clinica (CER), compresi quelli relativi alle procedure e ai dati, variano a seconda della classe del dispositivo e sono necessari per ottenere la certificazione CE dei dispositivi medici. I dispositivi a basso rischio di classe I possono effettuare l'autocertificazione CE. Al contrario, le altre classi di dispositivi (IIa, IIb, III) devono ottenere la certificazione del marchio CE tramite un organismo notificato (ON) accreditato. Il fabbricante deve presentare il fascicolo tecnico CE all'organismo notificato (NB) per la valutazione e il rilascio dell'approvazione del marchio CE e del certificato CE. Il rapporto di valutazione clinica (CER) per i dispositivi medici deve essere presentato insieme al fascicolo tecnico CE per soddisfare i requisiti di marcatura CE. Il CER deve essere aggiornato continuamente con nuove informazioni provenienti da controlli di sicurezza, studi di follow-up e gestione dei rischi.
Il Rapporto di Valutazione Clinica (CER) per i dispositivi medici è uno dei rapporti che devono essere presentati insieme al Fascicolo Tecnico CE per conformarsi ai requisiti CER.
Cos'è un Rapporto di Valutazione Clinica (CER)?
La redazione del rapporto di valutazione clinica include la valutazione e l'analisi dei dati clinici relativi a un Dispositivo Medico per verificarne la sicurezza e le prestazioni cliniche. La valutazione clinica dei dispositivi medici si basa sull'analisi completa dei dati clinici pre e post-commercializzazione pertinenti all'uso previsto. Il Rapporto di Valutazione Clinica include dati specifici per il dispositivo, nonché tutti i dati relativi ai dispositivi dichiarati equivalenti dal fabbricante.
Una relazione di valutazione clinica è costituita da letteratura scientifica e dati clinici analizzati, raccolti nell’ambito di una sperimentazione clinica del dispositivo in questione o dai risultati di altri studi su dispositivi sostanzialmente equivalenti; in tal caso, produttori avere pieno accesso ai dati tecnici, biologici e clinici del dispositivo equivalente e dimostrare chiaramente in che modo il proprio dispositivo sia conforme sotto tutti gli aspetti critici. La relazione di valutazione clinica (CER) di un dispositivo medico dimostra che il dispositivo raggiunge lo scopo previsto senza esporre gli utenti e i pazienti a ulteriori rischi.
EU MDR deve essere aggiornato ogni anno. La frequenza degli aggiornamenti del CER dipende dal livello di rischio ed è specifica per ciascun dispositivo. Se il dispositivo comporta rischi significativi, l'aggiornamento deve avvenire almeno una volta all'anno; se invece il dispositivo è commercializzato da un periodo di tempo significativo ed è ben consolidato, il CER può essere aggiornato ogni 2-5 anni. Eventuali modifiche apportate al progetto del dispositivo e qualsiasi nuova informazione derivante dai dati del sistema di monitoraggio post-commercializzazione (PMS) potrebbero richiedere un aggiornamento del rapporto CER.
La valutazione clinica dei dispositivi medici, come delineato nel Rapporto di Valutazione Clinica (CER), si basa sui fattori elencati di seguito.
- Letteratura scientifica attualmente disponibile; e/o
- Indagini cliniche effettuate; o
- Qualora la dimostrazione di conformità ai requisiti essenziali basata su dati clinici non sia ritenuta appropriata.
Fasi di redazione del Rapporto di Valutazione Clinica (CER)
In riferimento al nuovo Regolamento sui Dispositivi Medici dell'UE (MDR) – 2017/745, ci sono quattro diverse fasi per eseguire una valutazione clinica dei Dispositivi Medici al fine di preparare un Rapporto di Valutazione Clinica (CER) completo per l'MDR dell'UE.
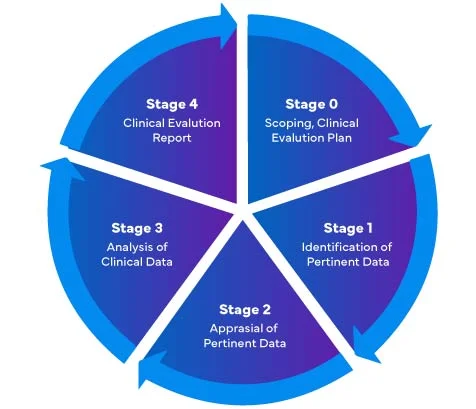
I produttori di dispositivi medici che entrano per la prima volta nel mercato dell'UE devono assicurarsi che il loro Rapporto di Valutazione Clinica sia conforme ai regolamenti EU MDR.
Freyr offre servizi di certificazione CE End-to-End ai produttori di dispositivi, inclusa la redazione del Rapporto di Valutazione Clinica in linea con i regolamenti EU MDR 2017/745 di nuova attuazione. Con una forte competenza regionale sui dispositivi medici dell'UE, Freyr soddisfa i requisiti specifici delle agenzie e personalizza di conseguenza il Rapporto di Valutazione Clinica.
Rapporto di valutazione clinica (CER)
- End-to-End supporto alla redazione di Rapporti di Valutazione Clinica, inclusa la ricerca bibliografica, secondo la revisione 4 del documento MEDDEV 2.7/1 e le linee guida del Regolamento sui Dispositivi Medici (MDR) dell'UE.
- Elaborazione di un piano di valutazione clinica per la vostra organizzazione.
- Identificare, ricercare, analizzare e assemblare la letteratura scientifica appropriata e applicabile.
- Sviluppare un modello di rapporto di valutazione clinica per la vostra organizzazione.
- Analisi delle lacune per il Rapporto di Valutazione Clinica esistente.
- Strumento DMS per il tuo team per contribuire collettivamente alla stesura del Rapporto di Valutazione Clinica.
- Integrare i dati PMS.
- Sviluppare una procedura operativa standard per il team per raccogliere i dati PMS e aggiornare i Rapporti di Valutazione Clinica.
- Gestione degli aggiornamenti periodici dei Rapporti di Valutazione Clinica esistenti, secondo le linee guida del MDR UE.
- Supporto dati PMS per i dispositivi esistenti sul mercato.
- Conformità alla Marcatura CE e servizi di Marcatura CE.

- Conformità garantita alle recenti normative applicabili.
- Team di esperti clinici qualificati.
- Contributi interfunzionali da esperti di dispositivi medici per conformarsi ai requisiti.
- Servizio completo che include conformità, revisione e pianificazione.

Domande frequenti (FAQ)
01. Che cos'è un rapporto di valutazione clinica (CER) e perché è importante?
Una relazione di valutazione clinica (CER) è un documento scientifico normativo che valuta sistematicamente tutte le prove cliniche disponibili al fine di dimostrare che un dispositivo medico è sicuro, funziona come previsto e offre un rapporto rischi/benefici clinico accettabile per l'uso previsto ai sensi del EU MDR. Essa svolge un ruolo centrale nella valutazione della conformità e nel mantenimento della conformità.
02. Quando occorre redigere e aggiornare un CER?
La preparazione del CER inizia nelle prime fasi dello sviluppo del prodotto, nell'ambito di un processo di valutazione clinica pianificato, e deve essere aggiornata regolarmente per tutto il ciclo di vita del dispositivo ogni volta che emergono nuove evidenze cliniche, dati post-commercializzazione o modifiche al profilo di rischio, garantendo che la valutazione del rapporto rischi/benefici rimanga aggiornata.
03. Quali sono gli elementi fondamentali che un CER conforme deve contenere?
Una CER solida dovrebbe riflettere una valutazione strutturata dei dati clinici pertinenti, confronti con le tecnologie all’avanguardia, un’analisi dei rischi e dei benefici, i risultati della sorveglianza post-commercializzazione e conclusioni oggettive sulla conformità ai requisiti EU MDR e prestazione previsti dal EU MDR .
04. In che modo lo “stato dell’arte” influisce su una relazione di valutazione clinica?
Il concetto di «stato dell'arte» costituisce il punto di riferimento della pratica clinica e della tecnologia consolidate, rispetto al quale vengono valutate le evidenze cliniche; esso garantisce che i benefici e i rischi del dispositivo siano contestualizzati rispetto agli standard attuali della medicina, favorendo un'interpretazione significativa delle evidenze.
05. È richiesto un CER per tutti i dispositivi medici ai sensi del EU MDR?
Sì, i CER sono obbligatori per tutti i dispositivi medici commercializzati nell'UE ai sensi del MDR, indipendentemente dalla classe di rischio, poiché documentano le prove cliniche essenziali per dimostrare la conformità ai requisiti normativi in materia di sicurezza e prestazioni.
06. Cosa distingue un CER di alta qualità da una semplice relazione di conformità?
Un CER di alta qualità integra una metodologia completa di ricerca bibliografica, affermazioni cliniche chiare e in linea con l'uso previsto, una valutazione rigorosa dei dati e approfondimenti ponderati sulle prestazioni cliniche e sui rischi, andando oltre il semplice adempimento formale per riflettere una comprensione approfondita delle aspettative normative e del contesto clinico.
07. Perché Freyr è considerata un partner di riferimento per i servizi relativi alle relazioni di valutazione clinica (CER)?
Freyr è ampiamente riconosciuta per il suo approccio allo sviluppo di studi comparativi basati sull'evidenza (CER) incentrato sugli aspetti normativi, che combina EU MDR profonda EU MDR , una solida valutazione delle prove cliniche e strategie orientate al ciclo di vita del prodotto. I suoi team multidisciplinari integrano le conoscenze cliniche, normative e relative alla fase post-commercializzazione per fornire studi CER scientificamente rigorosi, in grado di superare il vaglio degli organismi notificati e di garantire la conformità a lungo termine.







