
Servizi di conformità EU MDR - Panoramica
Il Regolamento sui Dispositivi Medici dell'UE (MDR) è entrato in vigore il 26 maggio 2021, dopo un periodo di transizione di tre anni e un'ulteriore estensione di un anno dovuta alla pandemia di COVID-19. I dispositivi immessi ora sul mercato dell'UE devono rispettare queste normative ed essere certificati CE secondo l'EU MDR dagli Organismi Notificati accreditati. I dispositivi già certificati CE secondo l'EU MDD godono invece di un periodo di grazia prima di dover conformarsi pienamente ai requisiti dell'EU MDR. Durante questo periodo, i dispositivi certificati sia sotto EU MDD che sotto EU MDR coesisteranno sul mercato con pari status e senza discriminazioni. Freyr offre servizi di conformità EU MDR ineguagliabili per aiutare le aziende di dispositivi medici a soddisfare i requisiti dell'EU MDR in modo tempestivo.
Tempistiche di Transizione e Nuove Classificazioni dei Dispositivi
Il Regolamento Europeo sui Dispositivi Medici (MDR) sarà pienamente efficace in tutti i Member States dell'EU e negli Stati dell'Associazione Europea di Libero Scambio (EFTA) a partire da maggio 2021 e fornisce ai produttori un periodo di transizione di 4 anni per la certificazione MDR completa dell'EU.
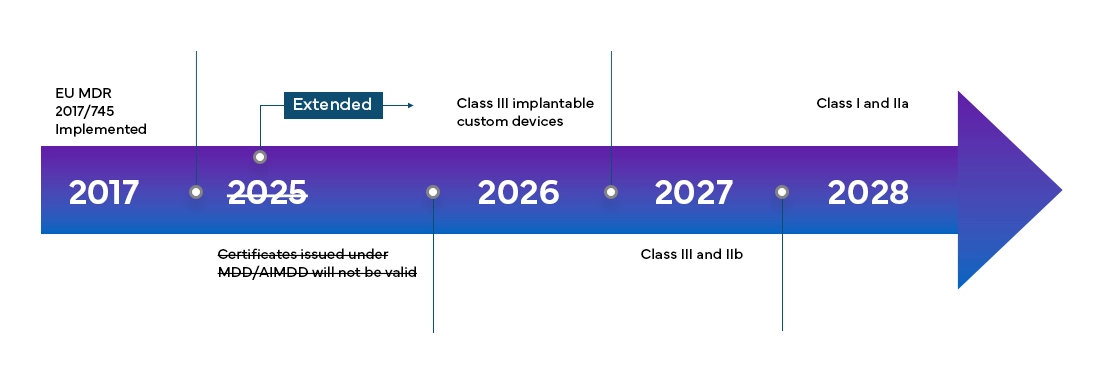
Il nuovo Regolamento Europeo sui Dispositivi Medici (MDR), come osservato, ha anche introdotto modifiche al sistema di classificazione dei dispositivi esistente, come:
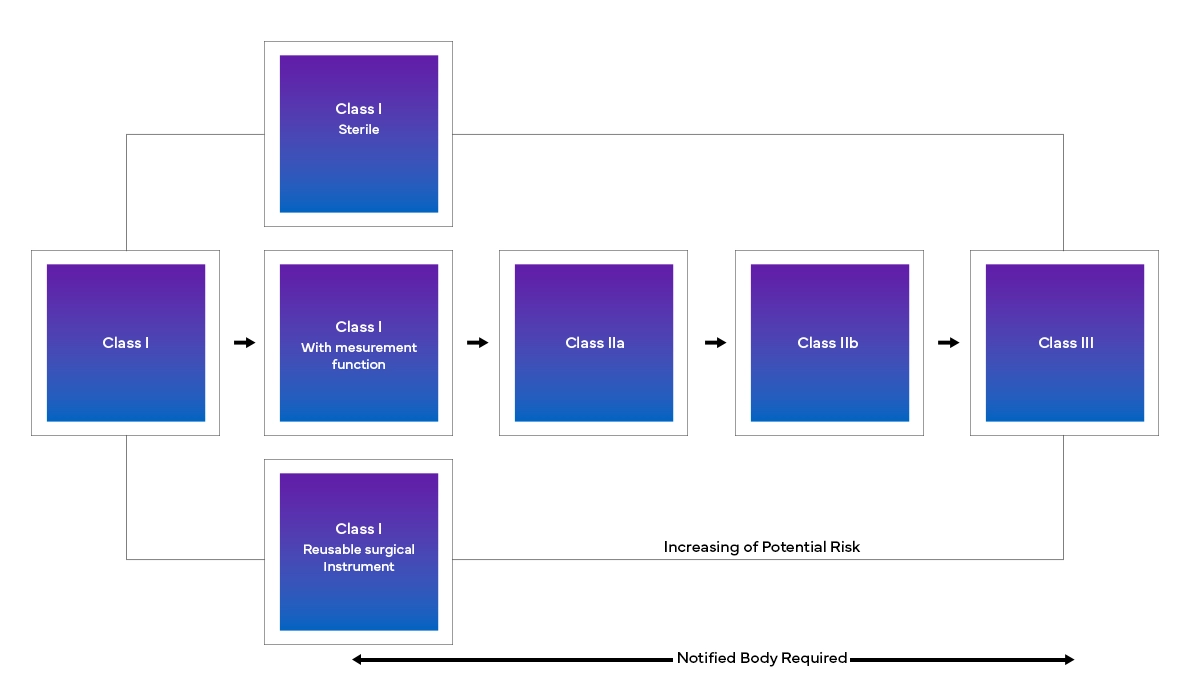
Dall'identificazione delle modifiche esatte da apportare all'implementazione in tempo reale, i produttori potrebbero dover affrontare una serie di sfide per conformarsi ai requisiti dell'EU MDR. Dalla decodifica della nuova struttura, alla classificazione accurata di un dispositivo, alla raccolta e presentazione di tutti i dati, sarà necessario un approccio regolatorio più dettagliato e trasversale per i produttori, per far fronte al nuovo Regolamento Europeo sui Dispositivi Medici. Con una rigorosa analisi delle lacune, Freyr assiste i clienti con lo status quo e fornisce le azioni regolatorie necessarie per la transizione e la Conformità all'EU MDR.
Servizi di conformità EU MDR
- Sviluppare una chiara strategia di implementazione del Regolamento sui Dispositivi Medici (MDR)
- Comprendere la nuova legislazione, condurre un'analisi delle lacune rispetto agli attuali Sistemi di Gestione della Qualità (QMS) e ai processi in atto.
- Sviluppare un piano dettagliato con un approccio interfunzionale per determinare gli aspetti del sistema qualità che richiederanno modifiche in conformità con il nuovo Regolamento sui Dispositivi Medici dell'UE.
- Formare più team per analizzare l'ambito del prodotto, la classificazione, la gestione del QMS ecc., all'interno dell'organizzazione, con un unico punto di contatto in ogni team
- Allocazione e pianificazione delle risorse
- Considerando l'interazione del vostro QMS con altre normative e utilizzando questa opportunità per ottimizzare i processi, pur consentendo flessibilità per incorporare futuri cambiamenti.
- Analizzare i dati di test disponibili e verificare eventuali requisiti aggiuntivi stabiliti dal MDR
- Coordinare le aspettative e il piano di transizione con i vostri Organismi Notificati dell'UE.
- Analisi delle Lacune per i Dispositivi Medici esistenti dalla Direttiva EU MDD ai Regolamenti EU MDR
- Supporto End-to-End per lo sviluppo del Rapporto di Valutazione Clinica (CER), inclusa la ricerca bibliografica secondo le linee guida del Regolamento Europeo sui Dispositivi Medici (EU MDR)
- Servizi End-to-End per i rapporti di sorveglianza post-commercializzazione (PMSR), i rapporti periodici di aggiornamento sulla sicurezza (PSUR) e il riepilogo della sicurezza e delle prestazioni cliniche (SSCP)
- Aumento delle risorse normative con opzioni di implementazione sia in sede che all'estero
- Servizi di Rappresentante Autorizzato Europeo (EAR)
- Conformità al Regolamento MDR e assistenza per la presentazione agli organismi notificati
- Intelligence normativa che copre il processo di importazione dei diversi mercati regolamentati
- Conformità al SGQ e audit simulati
- Sistema di gestione documentale e strumento per le aziende MDR
- Classificazione e riclassificazione dei dispositivi in base al rischio
- Implementazione UDI e consulenza.
- Servizi di sorveglianza post-commercializzazione conformi al Regolamento UE sui Dispositivi Medici
- Consulenza sulla gestione del rischio ISO 14971
- Formazione interna e online
- Persona responsabile per i servizi e l'assistenza di conformità normativa
- Identificazione degli Organismi Notificati MDR

Per un supporto normativo End-to-End sull' EU MDR, contattate Freyr







