
Panoramica sui prodotti combinati per dispositivi medici
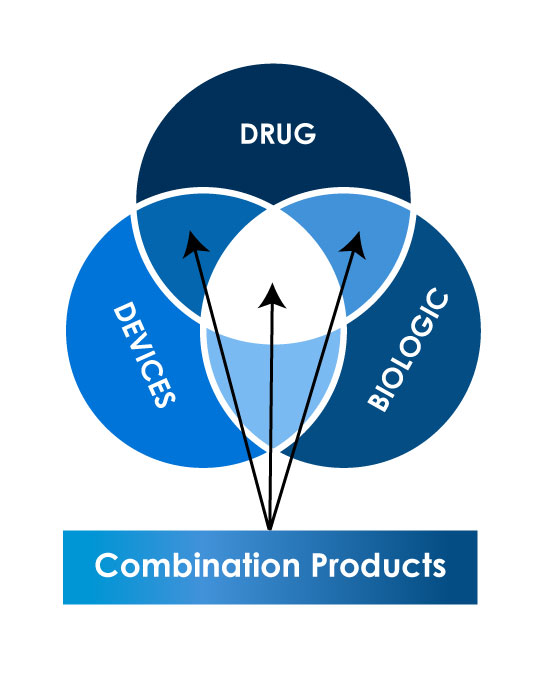
Nel dinamico mondo della sanità e dell'innovazione, i prodotti combinati medico-farmaceutici sono diventati un solido ponte che collega prodotti farmaceutici, dispositivi medici e biologici. Il mercato dei prodotti combinati è in rapida crescita, con un tasso di crescita annuale composto (CAGR) previsto dell'8,9%, dal 2023 al 2030. Il settore dei prodotti combinati farmaco-dispositivo è pronto per una crescita sostenuta, sostenuto dai progressi tecnologici, dal miglioramento delle infrastrutture sanitarie, da percorsi normativi più agevoli, da collaborazioni strategiche e da un impegno per l'assistenza centrata sul paziente.
Diverse tipologie di prodotti combinati
Scenario normativo globale per la registrazione dei prodotti combinati
L'interpretazione di ciò che costituisce un prodotto combinato può differire da una nazione all'altra, aumentando le complessità della registrazione di tali prodotti in vari paesi. Inoltre, le richieste e le procedure normative per i prodotti combinati possono presentare variazioni in termini di documentazione, comunicazione e convalida. Il panorama normativo per la registrazione dei prodotti combinati può differire significativamente a livello mondiale. Ecco le principali Autorità Regolatorie che supervisionano questi dispositivi a livello globale.
| Paese | Agenzia | Centri principali per l'approvazione |
|---|---|---|
| USA | Ufficio dei Prodotti Combinati (OCP) | Centro per la Valutazione e la Ricerca sui Farmaci (CDER) |
| Centro per la Valutazione e la Ricerca sui Prodotti Biologici (CBER) | ||
| Centro per i Dispositivi e la Salute Radiologica (CDRH) | ||
| UE | Organismi Notificati (ON) | Autorità Competente Nazionale (prodotti medicinali) |
| Organismi Notificati (ON) (dispositivi medici) | ||
| Giappone | Divisione Valutazione e Licenze o l'Ufficio Prodotti a base di Dispositivi Medici/Cellulari e Tessuti del Bureau per la Sicurezza Farmaceutica e Alimentare | Direttore della Divisione Valutazione e Licenze (DMDL), Ufficio per la Sicurezza Farmaceutica e Alimentare, Ufficio per la Sicurezza Farmaceutica e Medica, Ministero della Salute e del Welfare |
| Cina | Centro per l'Amministrazione della Standardizzazione dei Dispositivi Medici (CMDSA) | Centro per la Valutazione dei Dispositivi Medici (CMDE) |
| Centro per la Valutazione dei Farmaci (CDE) | ||
| Malesia | Agenzia Regolatoria Farmaceutica Nazionale | Agenzia Nazionale di Regolamentazione Farmaceutica (NPRA) |
| Agenzia per i dispositivi medici |
La registrazione dei prodotti combinati nei mercati internazionali richiede un approccio personalizzato, che implica una stretta collaborazione con le Agenzie Sanitarie competenti per l'approvazione. Il processo tipico per la registrazione dei prodotti combinati prevede i seguenti passaggi:
- Valutare se un dispositivo specifico soddisfa i criteri per la classificazione come prodotto combinato.
- Classificazione dei dispositivi in base ai rischi associati.
- Identificazione degli standard pertinenti e dei prerequisiti sui dati specificati dalla rispettiva Autorità Sanitaria.
- Generazione dei dati necessari come richiesto dall'Agenzia.
- Compilazione di un fascicolo tecnico in conformità ai requisiti specifici di ciascun paese.
- Presentare la domanda e rispondere a eventuali domande o preoccupazioni fino all'ottenimento dell'approvazione.
- Gestire il ciclo di vita del dispositivo dopo l'approvazione.
Le Nostre Competenze
- Analisi iniziale dei rischi
- Ricerche di mercato - Approfondimenti di mercato specifici per prodotto
- Rafforzamento dell'organico
- Bozza della strategia normativa
- Mercati e percorsi potenziali
- Fascicolo di progettazione e analisi dei rischi
- Sistema di Gestione della Qualità (SGQ) ISO 13485
- Programma di Audit Unico per Dispositivi Medici (MDSAP)
- Pre-valutazione QMS ISO 13485
- Strategia Regolatoria
- Freyr IMPACT (Piattaforma di Regulatory Intelligence)
- Verifica e validazione della progettazione
- Gestione del rischio
- Bozza della documentazione tecnica
- Strategia Regolatoria
- Requisiti normativi
- Strumento rDMS di Freyr (Sistema di Gestione Dati/Documentazione)
- Validazione di processo e clinica
- Etichettatura finale e Artwork
- Rappresentanza nel Paese
- Presentazione regolatoria
- La marcatura "Conformité Européenne" (CE) dell'Unione Europea (UE) e la marcatura UK Conformity Assessment (UKCA)
- Certificazione per l'accesso al mercato globale
- Supporto per gli audit dell'Organismo Notificato (ON)/Organismo Approvato
- Rappresentanza nel Paese
- Approvazioni regolatorie
- Sorveglianza post-commercializzazione (PMS)
- Sorveglianza Clinica Post-Commercializzazione (PMCF)
- Manutenzione annuale del fascicolo tecnico (CER/Gestione del rischio)
- Rinnovi Regolatori
- Nuovi lanci sul mercato
- Comunicazione con Autorità Competente, Organismo Notificato o Approvato
- Soluzioni automatizzate di Farmacovigilanza (FV)
Perché Freyr?
Registrazione dei Dispositivi Medici
- Strategia normativa completa per prodotti combinati.
- Supporto regolatorio per i documenti di sviluppo del prodotto, come i Design History Files (DHF).
- Strategia di conformità QMS.
- Conformità normativa, analisi delle lacune e risoluzione per documenti tecnici e sistemi di qualità.
- Servizi di etichettatura regolatoria e di redazione tecnica.
- Servizi di intelligenza normativa e di mercato.
- Servizi di traduzione di documenti ed etichettatura.
- Interazione e servizio con l'Agenzia Sanitaria.
- Servizi di artwork normativo.
- Servizi di Farmacovigilanza e sorveglianza post-commercializzazione.
- Servizi di pubblicazione.
- Servizi di redazione medica.

- Presentazioni riuscite per varie classi di IVD.
- Personale dedicato ed esperto per fornire supporto normativo per dispositivi medici e IVD.
- Presentazione puntuale dei documenti.
- Accesso dell'affiliata locale per affrontare le sfide dell'Autorità e i requisiti linguistici specifici.
- Supporto del rappresentante locale o legale con un modello economicamente vantaggioso.
- Gestione delle risorse normative/servizi di potenziamento del personale.








